Guide pratique : Le syndrome de Cornelia de Lange
Rédigé par Margot COMEL interne de Génétique Médicale, et Pr GENEVIEVE David responsable du Centre de référence des anomalies du développement syndromes malformatifs, CHU Montpellier
Relecture : Dr Olivier PATAT, responsable Centre de compétence des anomalies du développement et syndromes malformatifs CHU de Toulouse, Thomas EDOUARD responsable du Centre de référence des maladies endocriniennes rares de la croissance et du développement, Madame Catherine BRUSSET de L’Association Française Syndrome Cornelia de Lange
Qu’est-ce que le syndrome de Cornelia de Lange ?
Le syndrome de Cornelia de Lange (CdLS) (ORPHA: ; MIM #122470, #300590, #300882, #614701, #610759) est une maladie génétique rare, qui peut être due à des variants pathogènes dans plusieurs gènes différents, appartenant au groupe des cohésinopathies, c’est-à-dire une maladie génétique causée par un défaut dans le fonctionnement du complexe cohésine ou de ses protéines régulatrices (cf. infra aspects génétique). Il a été décrit pour la première fois en 1933 par la pédiatre hollandaise Cornelia de Lange. Sa prévalence est estimée entre 1/10 000 et 1/30 000 naissances.
Ce syndrome se caractérise par un phénotype très variable, allant de formes sévères typiques à des présentations plus légères parfois difficiles à identifier. Cette variabilité existe même au sein d’une même famille, selon le gène concerné.
Caractéristiques cliniques
Le syndrome associe habituellement un retard de croissance pendant la grossesse (détectable à l’échographie entre 20 et 25 semaines d’aménorrhée) et après la naissance. Le périmètre crânien est souvent plus petit que la normale (microcéphalie). Des courbes de croissance spécifiques au CdLS ont été développées et sont disponibles sur le site www.cdlsusa.org (filles et garçons).
Les cheveux sont généralement abondants, et s’étendent sur les tempes. Les cils et les sourcils sont particulièrement fournis. On retrouve également beaucoup de poils sur le reste du corps (hirsutisme).
Les anomalies des membres sont fréquentes et souvent asymétriques. Dans les formes les plus sévères, les avant-bras et les doigts peuvent être absents (oligodactylie). Plus couramment, les mains sont plutôt petites, avec des doigts courts avec parfois une limitation de l’extension des coudes. Concernant les jambes, les atteintes sont généralement moins sévères. Les pieds sont petits, les orteils peuvent être fusionnés et des hallux valgus sont retrouvés (déviation du gros orteil vers l’intérieur). Au niveau vertébral, la scoliose est retrouvée chez environ un tiers des patients et apparait vers 10 ans.
Les difficultés d’alimentation sont quasi constantes les premières années de vie, notamment dues à des difficultés d’acquisition de la succion-déglutition chez le nourrisson et de l’alimentation en morceaux. Le reflux gastro-œsophagien est présent chez la très grande majorité des patients et peut être observé tout au long de la vie. Non traité, il peut entrainer des complications. Les symptômes peuvent inclure des vomissements, une irritabilité, des douleurs, des troubles du sommeil et des infections respiratoires à répétition. Des troubles de l’oralité alimentaires sont également décrits, par hypersensibilité et refus de certaines textures. La sélectivité alimentaire peut persister dans le temps. La constipation est également fréquente (environ la moitié des patients). D’autres malformations digestives sont possibles mais moins communes (malrotation intestinale, sténose du pylore, hernie diaphragmatique congénitale).
Environ 25 à 30% des personnes malades ont une cardiopathie congénitale (malformation cardiaque), notamment un rétrécissement de la valve pulmonaire (le plus fréquent), une communication entre les ventricules ou les oreillettes, une coarctation de l’aorte.
Au niveau ophtalmologique, une paupière tombante (ptosis) est retrouvée chez plus de la moitié des individus et elle peut nécessiter une chirurgie si elle gêne la vision ou la marche. Une inflammation des paupières (blépharite) est fréquente également et nécessite une bonne hygiène. Les individus avec un CdLS sont souvent myopes et/ou astigmates.
Au niveau ORL, la surdité est fréquente, souvent bilatérale et présente dès l’enfance. Elle peut être due à des malformations de l’oreille moyenne et interne notamment, mais peut également être liée à des otites séro-muqueuses ce qui permet une amélioration de la surdité avec le temps. Les otites à répétition et les sinusites sont fréquentes (favorisées par le reflux gastro-oesophagien). Une séquence de Pierre Robin (malformation buccale avec micrognathie, rétrognatisme, glossoptose +/- fente palatine associée) ou une fente palatine isolée peuvent être retrouvées.
Les anomalies dentaires sont également décrites dans ce syndrome, avec une tendance au retard d’éruption des dents définitives, un encombrement et des malpositions dentaires. Il existe un risque accru de caries lié aux difficultés de brossage, à l’alimentation et au reflux gastrique.
Au niveau du système urinaire et génital, les anomalies rénales concernent environ 1 personne sur 10, avec des reflux vésico-urétraux et des malformations rénales. Chez les garçons, les testicules peuvent ne pas être descendus (cryptorchidie dans 73% des cas), l’urètre peut être encore ouvert à la naissance (hypospade dans 9% des cas) et les organes génitaux sont généralement de petite taille (57% des cas). Chez les filles, une malformation utérine (utérus bicorne) peut être retrouvée.
Les enfants avec un CdLS ont souvent un retard du neurodéveloppement et des troubles du comportement (auto-agressivité, comportements répétitifs, anxiété, mutisme), avec une tendance assez constante au retard de langage. La compréhension verbale est souvent meilleure que l’expression orale. Ils peuvent être accompagnés d’un trouble du spectre de l’autisme (TSA) et/ou d’épilepsie (1/4 des personnes). Les troubles du sommeil sont fréquents (difficultés d’endormissement et réveils nocturnes). La moitié des enfants marchent avant 24 mois, et la quasi-totalité avant l’âge de 10 ans dans les formes légères et modérées de la maladie.
D’autres manifestations à mentionner sont un possible déficit immunitaire pouvant se caractériser par des infections à répétition, des anomalies des lignées sanguines (anémie, thrombopénie) et un cutis marmorata (aspect marbré de la peau).
La puberté est généralement un peu retardée, avec un âge moyen de 13 ans chez les jeunes filles et 15 ans chez les jeunes garçons. Pendant la période pubertaire, le poids est également à surveiller car on remarque des situations de surpoids et d’obésité à cet âge. Les troubles de l’humeur sont également à rechercher, notamment une dépression et des difficultés au passage à l’âge adulte.
Concernant l’espérance de vie, elle n’est pas significativement réduite en l’absence de malformations sévères. L’amélioration de la prise en charge de ces personnes a permis d’améliorer le pronostic de cette maladie.
Aspects génétiques
Le CdLS est une maladie génétique, c’est-à-dire causée par une anomalie pathogène dans un gène. En termes médicaux, une anomalie dans un gène est appelée « variant pathogène » (auparavant mutation).
Différents gènes peuvent être impliqués dans cette maladie, bien qu’habituellement un seul soit en cause chez chaque individu atteint.
En fonction du gène atteint, la maladie peut être de transmission autosomique dominante (NIPBL la plupart du temps, SMC3, RAD21, BRD4) ou dominante liée à l’X (SMC1A, HDAC8). Ces gènes sont impliqués dans l’architecture et la régulation d’une protéine spécifique, la cohésine. C’est pourquoi on parle de cohésinopathie.
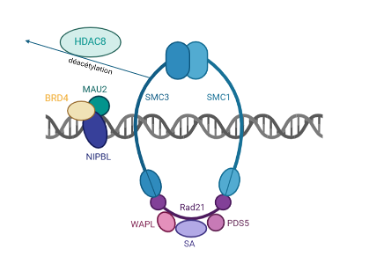
Schéma du complexe cohésine. L’anneau de cohésine est principalement formé par les protéines SMC1 et SMC3, ainsi que RAD21 et une protéine de la famille SA (STAG ou SCC3). Plusieurs facteurs régulent l’ouverture, la fermeture et la localisation de l’anneau : NIPBL et le complexe MAU2/BRD4, WAPL et PDS5. HDAC8 est une enzyme qui effectue une déacétylation de SMC3, essentielle à la stabilité du complexe cohésine.
Le terme « autosomique » signifie que les personnes atteintes peuvent aussi bien être des femmes que des hommes, la maladie n’est pas liée au sexe. Le terme « dominant » signifie qu’il suffit qu’une seule copie d’un des gènes impliqués dans ce syndrome soit altérée pour développer la maladie.
Pour les personnes atteintes du CdLS de transmission autosomique dominante (le cas plus fréquent), lorsqu’elles souhaitent avoir des enfants, elles ont 50% de risques de leur transmettre l’anomalie, quel que soit le sexe, et donc 50% de chances de ne pas leur transmettre.

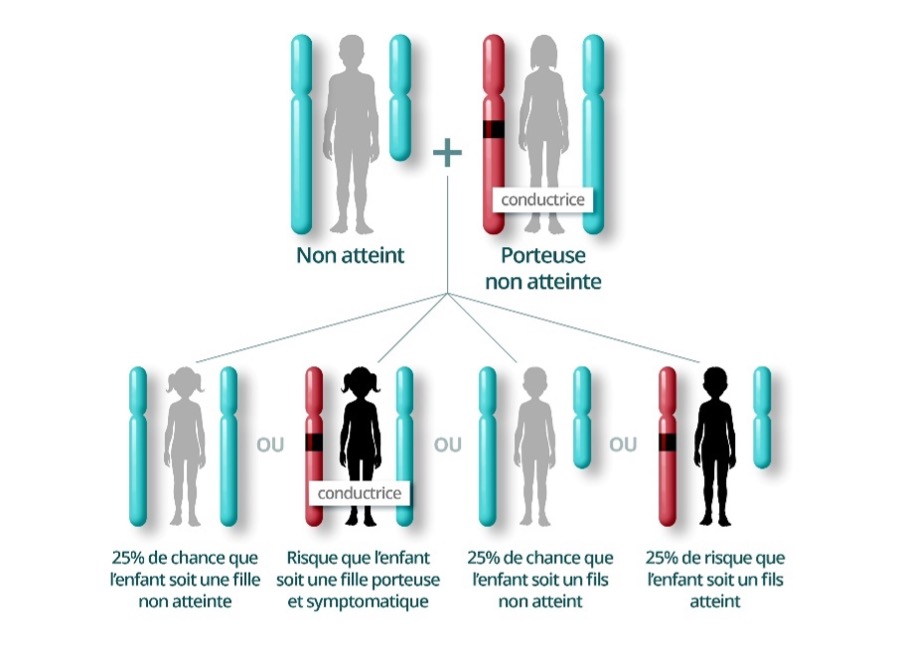
Le terme « dominante lié à l’X » signifie que chez une femme, la présence d’un seul chromosome X porteur de la variation pathogène suffit à entrainer des symptômes, avec une expression parfois variable en raison d’un phénomène d’inactivation du chromosome X. Les femmes atteintes auront 50% de risque de transmettre la variation génétique à leur descendance. Chez un homme, la présence d’un chromosome X porteur d’un variant pathogène entraine généralement une atteinte plus marquée, car il ne dispose pas d’un second chromosome X sain pour compenser. Les hommes atteints transmettront la variation génétique à toutes leurs filles, et à aucun de leurs fils.
Dans la grande majorité des cas, il s’agit d’une anomalie génétique « de novo ». Cela signifie que ni la mère, ni le père ne sont porteurs de cette anomalie et qu’elle est apparue accidentellement, probablement lors de la formation de l’ovocyte ou du spermatozoïde qui ont permis la naissance de l’enfant.
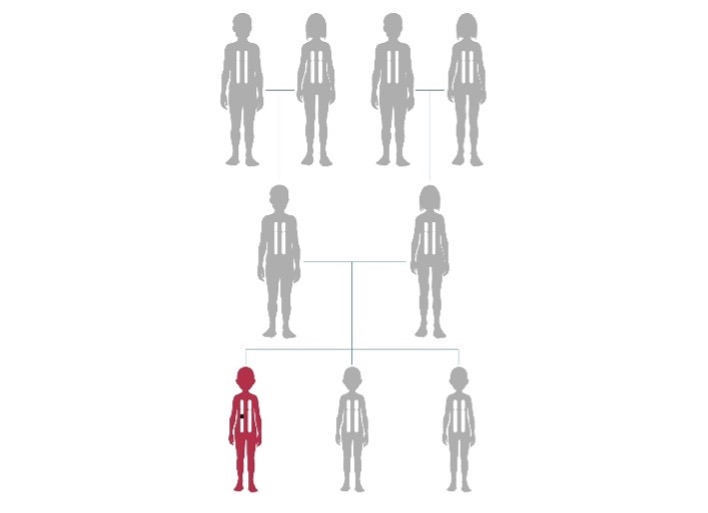
Cela implique pour les parents que le risque d’avoir un deuxième enfant atteint de ce syndrome est très faible. Cependant, ce risque n’étant jamais égal à zéro, il peut être proposé aux parents un diagnostic prénatal invasif ou non-invasif pendant la grossesse, si l’anomalie familiale est connue.
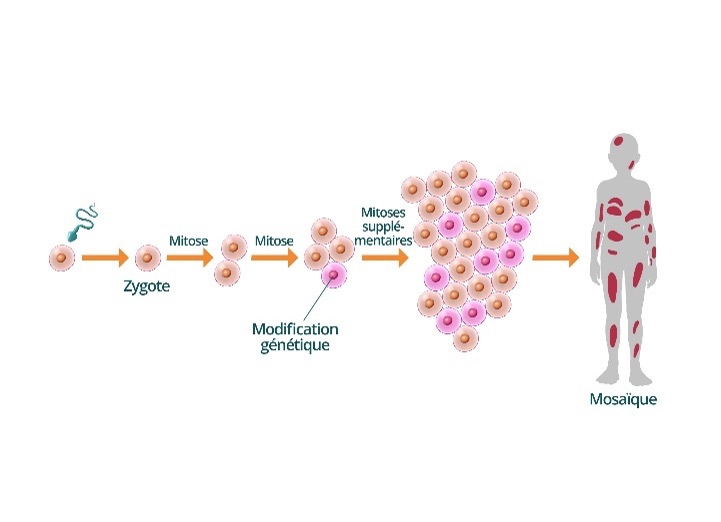
Dans certains cas (10 à 15%), l’anomalie génétique peut ne pas être présente dans toutes les cellules de l’enfant, mais uniquement dans une partie : c’est ce qu’on appelle une mosaïque, ce qui est parfois observé dans le syndrome CdL. Le variant génétique est présent dans certains tissus et pas dans d’autres. Il peut être absent du sang mais présent dans d’autres cellules. C’est pourquoi, si l’analyse du sang est normale mais que le diagnostic reste très probable, on peut proposer d’autres prélèvements comme la peau ou la salive.
Diagnostic
Le diagnostic peut être clinique si le médecin est habitué à voir des maladies rares, devant l’association de plusieurs signes caractéristiques. Mais aujourd’hui, ce diagnostic est majoritairement biologique.
Il repose sur l’identification d’un variant pathogène dans l’un des gènes impliqués dans ce syndrome, par analyse moléculaire (prise de sang, parfois complétée par d’autres prélèvements). Selon les études, entre 3 à 30% des personnes avec des signes cliniques de CdLS n’ont pas de variant pathogène identifié dans les gènes actuellement connus.
Le CdLS peut être suspecté pendant la grossesse en cas de signes échographiques évocateurs, comme le retard de croissance, les anomalies des membres, les malformations cardiaques ou digestives. Un diagnostic moléculaire est alors possible par prélèvement de villosités choriales ou par amniocentèse.
Prise en charge globale, traitement et suivi
Il n’existe pas à ce jour de traitement curatif du syndrome de Cornelia de Lange, la prise en charge est donc symptomatique et préventive. Elle vise à dépister et traiter les complications, optimiser le développement et l’autonomie, et améliorer la qualité de vie de l’enfant et de sa famille.
La prise en charge doit se faire en lien avec un Centre Expert Maladies Rares (Centre de Référence Maladies Rares, CRMR, ou Centre de Compétence Maladies Rares, CCMR) du CdLS qui sont les CRMR/CCMR des anomalies du développement et syndromes malformatifs ou les CRMR/CCMR des maladies osseuses constitutionnelles des CHU de Montpellier et de Toulouse (coordonnées dans la partie « Professionnels de santé en Occitanie »). Les filières de santé maladies rares concernées sont AnDDI-Rares et OSCAR.
L’équipe multidisciplinaire spécialisée nécessite différents spécialistes : des généticiens cliniciens, des pédiatres, des cardiologues, des gastro-entérologues, des urologues, des endocrinologues, des pneumologues, des orthopédistes, des médecins ORL, des dentistes, des ophtalmologues, des neurologues et des psychiatres, tous en lien avec le médecin traitant. Le soutien psychologique de l’enfant et de sa famille, ainsi que l’accompagnement socio-éducatif font partie intégrante du traitement. Les professionnels paramédicaux sont également impliqués dans cette prise en charge : kinésithérapeutes, psychomotriciens, orthophonistes, ergothérapeutes, orthoptistes, psychologues, éducateurs spécialisés, infirmiers d’éducation thérapeutique, diététiciens et assistants sociaux.
Scolarisation
La scolarisation des enfants porteurs d’un CdLS doit être pensée de façon individualisée en fonction du niveau de développement, des compétences de communication et d’autonomie, ainsi que des troubles associés (troubles du comportement, surdité, troubles visuels, motricité).
Une inclusion en milieu scolaire ordinaire est souhaitable lorsque cela est possible, avec la mise en place d’un accompagnement adapté (auxiliaire de vie scolaire/AESH, aménagements pédagogiques, temps de repos, matériel spécifique). Le recours à des supports de communication alternative ou augmentée peut considérablement favoriser l’accès au langage et aux apprentissages.
Pour un soutien à la scolarisation, l’intervention et l’accompagnement par un SESSAD peut également être envisagé.
Dans les situations où le retard du développement ou les troubles du comportement sont plus marqués, une orientation vers des dispositifs plus spécialisés comme les classes ULIS ou ceux relevant du médico-social (IME, ITEP) peut être proposée afin de garantir un environnement éducatif structuré et sécurisé. Les activités adaptées doivent être encouragées, notamment lors des séances de sport.
Une collaboration étroite entre l’équipe pédagogique, (enseignants, enseignants référents, médecin scolaire) les parents et l’équipe médico-sociale est essentielle pour favoriser l’intégration, l’évolution des compétences et le bien-être de l’enfant à l’école. Un projet personnalisé de scolarisation (PPS) est fortement recommandé pour organiser le parcours et coordonner les interventions.
Pour en savoir plus sur la scolarisation : https://www.maladies-rares-occitanie.fr/accompagner/vie-quotidienne/scolarite
Suivi médical et paramédical
Le diagnostic et la prise en charge du CdLS requièrent une approche multidisciplinaire essentielle, impliquant différents professionnels de santé dans des structures spécialisées, comme les Centres de Compétence et les Centres de Référence Maladies Rares.
Dès l’évaluation initiale, la collaboration entre les différents médecins spécialistes et le médecin ou pédiatre traitant est importante pour gérer les potentielles et différentes complications associées (anomalies rénales, cardiaques, vertébrales, etc.).
À cela s’ajoutent les interventions paramédicales qui sont également essentielles.
L’orthophonie est à débuter le plus tôt possible (dès l’identification des troubles de l’oralité) avec un travail sur la communication (orale et alternative), la mise en place d’outils de Communication Alternative et Augmentée (CAA) et un travail sur l’oralité alimentaire.
En ce qui concerne l’alimentation, un dépistage précoce des difficultés est fondamental (par un orthophoniste spécialisé dans la mesure du possible). Le reflux gastrique doit être traité (inhibiteurs de la pompe à protons). L’adaptation de la position après les repas et des textures sont également des pistes pour améliorer la qualité de vie liée à la nutrition. Enfin, des chirurgies peuvent être envisagées en cas d’échec.
La psychomotricité est nécessaire pour stimuler le développement moteur global, réaliser un travail sur le schéma corporel et l’équilibre, et la gestion des troubles sensoriels.
La kinésithérapie est utile pour la motricité globale et la prévention des complications orthopédiques.
L’ergothérapie permet le développement de l’autonomie dans les activités quotidiennes (habillage, repas, toilette), l’adaptation à l’environnement et la mise en place d’aides techniques.
Enfin, l’accompagnement psychologique est indispensable, pour l’enfant, les parents et la fratrie.
En cas de troubles du sommeil, des mesures d’hygiène peuvent être suffisantes (horaires réguliers, rituel de coucher apaisant, environnement calme, éviction des écrans). En cas d’échec de ces mesures, un traitement par mélatonine pourra être proposé.
Toute anomalie de croissance doit faire référer à un endocrinologue pédiatre (ralentissement de la vitesse de croissance, taille inférieure aux courbes de la population générale pour l’âge). Un éventuel traitement hormonal substitutif sera envisagé pour les mêmes indications que celles de la population générale. Une telle prise en charge globale permet d'optimiser le suivi à long terme et la qualité de vie des personnes, en leur permettant de développer leur autonomie, d’établir une bonne communication, de participer à la vie sociale et familiale et d’avoir des activités adaptées ainsi que des loisirs.
Pour les femmes atteintes d’un CdLS et souhaitant une grossesse, un suivi adapté peut être nécessaire en fonction des atteintes associées (cardiopathie notamment). Une consultation pré-conceptionnelle et un conseil génétique sont recommandés avant la grossesse. Les risques de transmission ont été abordés dans la partie « Aspects génétiques ». Le diagnostic prénatal et préimplantatoires sont possibles si le variant génétique pathogène est connu.
Accès aux droits
Des aides financières, telle que l’Allocation pour l’Éducation des Enfants en situation de Handicap (AEEH), peuvent également être mises en place pour compenser les frais liés à la situation de handicap pour les enfants de moins de 20 ans. Elle est versée aux parents par la CAF et nécessite une demande auprès de la MDA (MDPH)
L’AEEH est basée sur les éléments transmis par la famille (projet de vie, justificatifs de frais,…) et sur les évaluations des différents professioennels intervenant auprès de l’enfant : médicaux, paramédicaux, scolaires et médico-sociaux. Pour l’adulte atteint, d’autres aides sont disponibles comme l’Allocation aux Adultes Handicapés (AAH), la Prestation de Compensation du Handicap (PCH), la Reconnaissance de la Qualité de Travailleur Handicap (RQTH) et la reconnaissance des aidants familiaux.
Une Carte Mobilité Inclusion (CMI) peut également être obtenue.
Le médecin référent ou le médecin traitant se charge de remplir la demande de prise en charge à 100% au titre d’une Affection Longue Durée (ALD) hors liste auprès de la CPAM. Il remplit également le certificat médical pour le dossier de la Maison Départementale de l’Autonomie (MDA) (anciennement appelée Maison Départementale des Personnes en situation de Handicap, MDPH).
Des ressources spécifiques liées à la maladie rare, peuvent être joints aux différents dossiers de demande telles que la fiche focus handicap d’Orphanet (Fiche focus Handicap) et le formulaire complémentaire pour la MDA (https://www.monparcourshandicap.gouv.fr/sites/default/files/2023-11/CNSA_Formulaire_Information_compl%C3%A9mentaires_pour_la_MDPH_ou_la_MDA.pdf )
Enfin, le dispositif régional Maladies Rares Occitanie est également à la disposition des professionnels qui vous accompagnent dans les différentes démarches sociales : aide à la constitution du dossier pour la Maison Départementale de l’Autonomie (MDA), informations sur les droits, orientation vers les ressources locales, soutien aux aidants.
Pour en savoir plus sur les démarches d’accès aux droits : https://www.maladies-rares-occitanie.fr/accompagner/vie-quotidienne/acces-aux-droits
Associations de patients
- Association Française du Syndrome de Cornelia de Lange : https://afscdl.fr/
- Alliance Maladies rares : https://alliance-maladies-rares.org/
- CdLS World (association internationale) : www.cdlsworld.org
Références
- Protocole National de Diagnostic et de Soins (PNDS), Haute Autorité de Santé. Syndrome de Cornelia de Lange. https://www.has-sante.fr/jcms/p_3385210/fr/syndrome-de-cornelia-de-lange
- Deardorff MA, Noon SE, Krantz ID. Cornelia de Lange Syndrome. 2005 Sep 16 [updated 2020 Oct 15]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301283.
- Kline AD, Moss JF, Selicorni A, et al. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat Rev Genet. 2018;19:649-666.
Professionnels de santé en Occitanie Est
Centre de Compétences des maladies endocriniennes de la croissance et du développement
Hôpital Arnaud de Villeneuve
34295 Montpellier CEDEX 5
France
Centre de référence des anomalies du développement syndromes malformatifs (pour les formes complexes comme les formes avec anneau de l’X)
Hôpital Arnaud de Villeneuve
Site internet : https://maladies-rares.chu-montpellier.fr/fr/
34295 Montpellier CEDEX 5
France
Plateforme d’Expertise Maladies rares Montpellier-Nîmes (PEMR)
Hôpital Arnaud de Villeneuve
34295 Montpellier CEDEX 5
France
Professionnels de santé en Occitanie Ouest
Centre de référence des maladies endocriniennes rares de la croissance et du développement (CRESCENDO)
CHU de Toulouse - Hôpital Purpan
TSA 70034 – 31059 Toulouse Cedex 9
France
Centre de référence des pathologies gynécologiques rares (PGR)
CHU de Toulouse – Hôtel-Dieu Saint-Jacques
TSA 80035 – 31059 Toulouse Cedex 9
France
Centre de compétence des anomalies du développement et syndromes malformatifs
CHU de Toulouse - Hôpital Purpan
TSA 70034 – 31059 Toulouse Cedex 9
France
Plateforme d’Expertise Maladies rares Toulouse (PEMR)
CHU Toulouse
Direction de la recherche et l’innovation (DRI) - Hôtel Dieu