Guide pratique : Syndrome KBG
Qu'est ce que le syndrome KGB ?
Guide rédigé par : Valentin Ruault et Professeur David Geneviève Centre de référence Anomalies du développement CHU Montpellier
Dernière mise à jour février 2018
Le syndrome KBG a été décrit pour la première fois en 1975 et doit son nom aux initiales des trois premiers patients décrits.
Nous possédons tous 46 chromosomes. Vingt-trois nous viennent de notre mère, et 23 de notre père. Nous possédons donc un double de chacun de nos gènes.
Dans ce syndrome, une des copies du gène ANKRD11 (MIM *611192) est atteinte. L’autre est normale, mais ne suffit pas à compenser cette perte. C’est donc un gène dominant. Il est situé sur le chromosome 16.
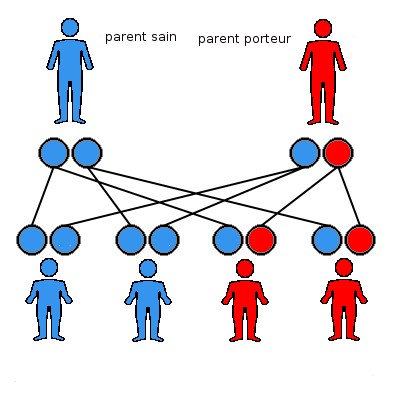
Lors de la conception d’un embryon, et donc d’un enfant, la maman donne un lot de 23 chromosomes, et le papa donne aussi un lot de 23 chromosomes. Puisque nous possédons tous deux lots de 23 chromosomes, il y a quatre combinaisons possibles de patrimoine génétique pour l’enfant, schématisées ci-dessus. Chaque rond représente un lot de chromosomes.
Dans le cadre d’une maladie dominante, le parent porteur de la mutation a donc un risque sur deux de transmettre la mutation (le lot de chromosomes rouge). L’enfant a donc un risque sur deux d’être porteur de la mutation, et donc de la maladie.
La protéine ANKRD11 est un régulateur de la transcription par modulation de la chromatine (donc régule l’ADN). Elle contrôle l’acétylation des histones et l’expression des gènes au cours du développement, notamment des neurones.
Le nombre de personnes touchées par ce syndrome est probablement sous-évalué et donc difficile à estimer. Cependant, le gène ANKRD11 semble être un des gènes les plus fréquemment mis en cause dans les déficiences intellectuelles et difficultés d’apprentissage de l’enfant.
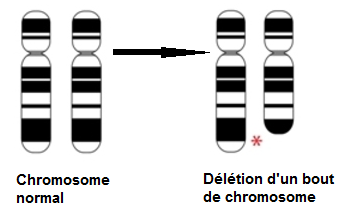
Un syndrome proche lui est rattaché : la délétion 16q24.3. En effet, lors de la délétion d’un bout du bras long d’un des deux chromosomes 16, on observe parmi les gènes délétés, ANKRD11.
Caractéristiques cliniques
Caractéristiques cliniques du syndrome KBG (avec entre parenthèses le pourcentage de patients touchés par le symptôme)
Il associe :
- un retard du développement et des apprentissages (90%, âge moyen de la marche 21 mois, premiers mots entre 2 et 3 ans)
- des difficultés d’apprentissage (lorsque le QI est compris entre 70 et 85) voire une déficience intellectuelle (lorsque le QI est inférieur à 70) de degré variable, rarement profonde (95% des enfants, résultats hétérogènes, environ 50% en école ordinaire, 50% en école spécialisée), pouvant conduire à une diminution de l’autonomie (40%).
- des manifestations neurologiques
- des troubles du comportement (50%)
- des troubles du sommeil (40%)
- des crises d’épilepsie de types variés, souvent peu sévères (40%)
- des troubles déficitaires de l’attention avec hyperactivité (10%)
- une hypoplasie du vermis (rare)
Il s’y ajoute d’autres symptômes, et quelques particularités morphologiques (voir ci-dessous).
critères diagnostiques
|
Un diagnostic de syndrome KBG doit être évoqué chez un patient présentant un retard du développement, des difficultés d’apprentissage, un retard de langage ou des troubles du comportement majeurs avec au moins deux critères majeurs ou un critère majeur et deux critères mineurs. |
|
|
Critères majeurs |
Pourcentage de patients concernés |
|
Macrodontie ou aspect de KBG de la dentition lactéale dans l’enfance |
67 - 85 % |
|
Taille inférieure au 10ème percentile |
40 % |
|
Otites moyennes aiguës récurrentes et/ou perte d’audition |
44 % |
|
Parent au 1er degré porteur d’un syndrome KBG |
10 - 20 % |
|
Critères mineurs |
|
|
Brachydactylie ou anomalie notable de la main |
50 - 60 % |
|
Crises d’épilepsie |
40 % |
|
Cryptorchidie |
40 % des garçons |
|
Trouble de l’alimentation |
20 - 30 % |
|
Anomalie du palais |
25 % |
|
Fontanelle antérieure large ou de fermeture tardive |
22% |
Adapté de la publication de Low and al., 2016
Il est bien entendu qu’aucun patient ne présente tous ces symptômes, et qu’on ne retrouve chez chacun qu’un nombre variable de ceux-ci.
Manifestations dentaires :
- de grandes incisives centrales supérieures de la denture définitive (67 - 85%), parfois de la denture déciduale, avec parfois fusion des incisives supérieurs centrales et latérales
Manifestations de la sphère ORL :
- une insuffisance vélaire (60%), un palais creux, parfois une fente du palais (10%)
- des otites moyennes aiguës récidivantes fréquentes, pouvant conduire à une perte d’audition voire à la surdité (27% de surdité, à un âge moyen de 4 ans lorsqu’elle est présente, et 65% d’appareillages auditifs)
- des troubles du langage (20%)
Manifestations ophtalmologiques :
- un strabisme
- une myopie (rare)
- une cataracte (rare)
Au niveau des extrémités :
- des doigts courts, en particulier le 5ème doigt (brachydactylie, 60%)
- une déviation du 5e doigt de la main (clinodactylie, 30%)
- des ongles dystrophiques (19%, également retrouvés sur les ongles des pieds)
Manifestations squelettiques :
- des anomalies costovertébrales (50%) telles que des côtes cervicales, des vertèbres de formes anormales voire fusionnées, parfois un spina bifida
- une fontanelle large, qui se ferme tard (22%)
- une dysplasie des hanches (16%) voire ostéochondrite primitive de hanche
- une ostéopénie précoce (15%) voire ostéoporose précoce
- une scoliose
- un retard de développement osseux
Manifestations endocrinologiques :
- une petite taille postnatale (40%) malgré des mensurations de naissance dans la moyenne
- une avance de l’âge de la puberté (15%)
Manifestations uro-génitales :
- une cryptorchidie chez le garçon (des testicules qui ne sont pas complètement descendus dans les bourses, 40%)
- une ectopie rénale (rare)
Manifestations digestives :
- des difficultés alimentaires du nouveauné (20%)
- un reflux gastroœsophagien (RGO)
- une constipation
- une malrotation des viscères (rare)
Manifestations cardiaques :
- des malformations cardiaques congénitales : communication interventriculaire, sténose de l’artère pulmonaire ou valvulopathies (15%)
Dans le cadre de la délétion 16q24.3, on note, en plus des symptômes cités ci-dessus, et en fonction du nombre de gènes perdus à cause de la délétion :
- des malformations cardiaques congénitales plus fréquentes (33%, liées à la perte des gènes ZFPM1 et ZNF778)
- un astigmatisme (30%, lié à la perte du gène ZFPM1)
- des malformations du système nerveux central (28%, liées à la perte du gène CDH15)
- une thrombocytopénie (un manque de plaquettes, 25%, liée à la perte du gène ZFPM1)
- des troubles du spectre autistique
- des malformations rénales plus fréquentes
Focus sur les compétences psychomotrices
Le QI moyen des patients atteints du syndrome KBG est globalement compris entre 60 et 80. Certains patients présentent donc une déficience intellectuelle, définie par un QI inférieur à 70, tandis que d’autres, qui ont un QI compris entre 70 et 85, ont des difficultés d’apprentissage.
Le QI verbal est habituellement meilleur que le QI performance.
Tous les enfants savent marcher, à un âge moyen de 21 mois, et aucune régression n’a été observée. Les enfants disent leurs premiers mots en moyenne à 36 mois.
Le niveau scolaire de ces enfants est variable. Certains ont fait des études supérieures.
Plus de la moitié des patients ont un emploi.
Cause
Il s’agit d’une affection autosomique dominante. Le syndrome KBG est parfois dû à des mutations de novo, c’est à dire accidentelles et non héritées, du gène ANKRD11. Dans cette situation, le risque pour un couple d’avoir un deuxième enfant atteint du syndrome KBG est proche de celui de la population générale (il n’y a pas encore eu de démonstration de mosaïque germinale dans cette affection).
Le risque pour un patient atteint, avec mutation dans le gène ANKRD11, d’avoir un enfant avec un syndrome KBG est par contre de 50%.
Diagnostic
Des critères diagnostiques ont été définis, à partir des différents symptômes listés précédemment. Si suffisamment de critères sont réunis, il peut être proposé d’étudier le gène ANKRD11 dans l’ADN contenu dans les globules blancs ou dans la salive. Il suffira alors de réaliser une prise de sang ou un frottis jugal, sans nécessité d’être à jeun.
Plus souvent, lorsque le tableau clinique n’est pas évocateur, on réalise une étude de tous les gènes du patient (un exome). On trouve alors une mutation dans le gène ANKRD11 qui explique la symptomatologie, et pose le diagnostic. On peut également trouver une délétion 16q24.3, grâce à un examen s’appelant ACPA (Analyse Chromosomique par Puces à ADN), qui explique elle aussi les symptômes.
Pronostic
La sévérité de la pathologie est variable. Parfois, l’autonomie des patients est possible.
Traitement
Aujourd’hui, il n’existe pas de traitement capable de guérir le syndrome KBG.
On peut en revanche prévenir ou traiter certains symptômes, avec par exemple :
- des antiépileptiques, contre les crises d’épilepsie ;
- de la mélatonine pour favoriser l’endormissement ;
- du methylphenidate contre les troubles du comportement et de l’attention ;
- des prothèses auditives, contre la perte d’audition ;
- des lunettes de vue, contre les troubles oculaires.
La chirurgie corrige le plus souvent les malformations. Citons par exemple :
- la chirurgie cardiaque, pour refermer une éventuelle communication interventriculaire;
- la chirurgie plastique infantile, pour refermer une fente labiopalatine ;
- la cure chirurgicale de cryptorchidie ;
- la chirurgie orthopédique, par exemple en cas de scoliose très sévère, ou de dysplasie des hanches sévère.
Des études sont actuellement en cours, afin d’évaluer l'intérêt des hormones de croissance dans le cadre du retard statural.
ATTENTION : la nature des appareillages auditifs rend souvent leur prise en charge par les MDPH difficile. En cas de non remboursement complet des dépenses vous devez impérativement demander de l’aide auprès d’une assistante sociale pour remplir votre dossier MDPH. Le Réseau Maladies Rares est à votre disposition pour vous conseiller en cas de difficultés pour les prises en charge et les remboursements des appareillages.
Surveillance
Le dépistage des anomalies suivantes doit être mis en œuvre par des équipes spécialisées.
Toute anomalie mise en évidence lors du bilan initial doit être prise en charge par l’équipe médicale spécialisée.
La surveillance générale sera programmée par le Généticien puis le Pédiatre ou le Médecin Traitant, avec la famille, en fonction des problèmes médicaux.
À la découverte du diagnostic
- examen clinique attentif avec recherche d’une fente labiopalatine, palpation testiculaire et examen des hanches
- consultation de cardiologie avec échographie cardiaque et ECG
- consultation de neuropédiatrie avec EEG
- échographie abdominorénale
- bilan auditif ORL
- bilan ophtalmologique
- bilan orthophonique
- bilan ergothérapique si nécessaire
- bilan kinésithérapique si nécessaire
Ne pas oublier :
- 100% ALD hors liste
- dossier MDPH
- orientation CAMSP si nécessaire
- PAI et/ou PPS
- discuter de l'intérêt d’une AVS
- soutien scolaire si nécessaire, en particulier français et mathématique
Dans l’enfance
- surveillance attentive du développement psychomoteur (prévoir un bilan psychomoteur du développement) et des acquisitions scolaires
- surveillance semestrielle de la croissance et bilan endocrinien de la croissance
- surveillance d’une puberté précoce
- bilan ophtalmologique annuel
- bilan auditif annuel
- séances d’orthophonie si nécessaire
- kinésithérapie si nécessaire
- ergothérapie si nécessaire
- bilan dentaire annuel
- orthodontie si besoin dès les premières dents définitives
- bilan phosphocalcique sanguin et urinaire : PTH, 25OHD, calcium, phosphate, PAL et rapport calciurie / créatininurie sur une miction à l’âge de 5 - 6 ans, à répéter à l’âge de 10 ans
- ostéodensitométrie à l’âge de 5-6 ans, à répéter à l’âge de 10 ans
- évaluation diététique des apports calciques à l’âge de 5-6 ans, à répéter à l’âge de 10 ans
- si infections ORL à répétition : dosage pondéral des immunoglobulines et NFS
À l’adolescence
- surveillance semestrielle clinique de l’apparition d’une scoliose
- surveillance semestrielle de la croissance et bilan endocrinien
- évaluation du développement et des acquisitions scolaires
- séances d’orthophonie si nécessaire
- kinésithérapie si nécessaire
- ergothérapie si nécessaire
- bilan ophtalmologique annuel
- bilan auditif annuel
- bilan dentaire annuel
- bilan phosphocalcique sanguin et urinaire : PTH, 25OHD, calcium, phosphate, PAL et rapport calciurie / créatininurie sur une miction à l’âge de 16 - 17 ans
- ostéodensitométrie à l’âge de 16 - 17 ans
- évaluation diététique des apports calciques à l’âge de 16 - 17 ans
À l’âge adulte
- ostéodensitométrie tous les cinq ans
- bilan phosphocalcique sanguin et urinaire : PTH, 25OHD, calcium, phosphate, PAL et rapport calciurie / créatininurie sur une miction tous les deux ans
- évaluation diététique des apports calciques
- bilan ophtalmologique annuel
- bilan auditif annuel
- bilan dentaire annuel
- séances d’orthophonie si nécessaire
- kinésithérapie si nécessaire
- ergothérapie si nécessaire
Il faut ajouter, en cas de Del16q24.3, une IRM cérébrale au moment du diagnostic et des numérations plaquettaires annuelles.
Références
N° MIM : #148050
N° ORPHA : 2332 pour KBG
261250 pour Del16q24.3
Herrmann J, Pallister PD, Tiddy W, Opitz JM. The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig Artic Ser. 1975;11: 7–18.
Tollaro I, v Bassarelli, Calzolari C, Franchini F, Giovannucci Uzielli ML, Vieri PL. [Dento-maxillo-facial anomalies in the KBG syndrome]. Minerva Stomatol. 1984;33: 437–446.
Fryns JP, Haspeslagh M. Mental retardation, short stature, minor skeletal anomalies, craniofacial dysmorphism and macrodontia in two sisters and their mother. Another variant example of the KBG syndrome? Clin Genet. 1984;26: 69–72.
Soekarman D, Volcke P, Fryns JP. The KBG syndrome: follow-up data on three affected brothers. Clin Genet. 1994;46: 283–286.
Rivera-Vega MR, Leyva Juárez N, Cuevas-Covarrubias SA, Kofman-Alfaro SH. Congenital heart defect and conductive hypoacusia in a patient with the KBG syndrome. Clin Genet. 1996;50: 278–279.
Devriendt K, Holvoet M, Fryns JP. Further delineation of the KBG syndrome. Genet Couns. 1998;9: 191–194.
Mathieu M, Helou M, Morin G, Dolhem P, Devauchelle B, Piussan C. The KBG syndrome: an additional sporadic case. Genet Couns. 2000;11: 33–35.
Smithson SF, Thompson EM, McKinnon AG, Smith IS, Winter RM. The KBG syndrome. Clin Dysmorphol. 2000;9: 87–91.
Iwasaki K. [KBG syndrome]. Ryoikibetsu Shokogun Shirizu. 2001; 34–35.
Dowling PA, Fleming P, Gorlin RJ, King M, Nevin NC, McEntagart M. The KBG syndrome, characteristic dental findings: a case report. Int J Paediatr Dent. 2001;11: 131–134.
Tekin M, Kavaz A, Berberoğlu M, Fitoz S, Ekim M, Ocal G, et al. The KBG syndrome: confirmation of autosomal dominant inheritance and further delineation of the phenotype. Am J Med Genet A. 2004;130A: 284–287.
Brancati F, D’Avanzo MG, Digilio MC, Sarkozy A, Biondi M, De Brasi D, et al. KBG syndrome in a cohort of Italian patients. Am J Med Genet A. 2004;131: 144–149.
Maegawa GHB, Leite JCL, Félix TM, da Silveira HLD, da Silveira HE. Clinical variability in KBG syndrome: report of three unrelated families. Am J Med Genet A. 2004;131: 150–154.
Davanzo AMG, Rosalia G, Biondi M, De Brasi D, Colucci AR, Panetta A, et al. Eight isolated cases of KBG syndrome: a new hypothesis of study. Eur Rev Med Pharmacol Sci. 2005;9: 49–52.
Brancati F, Sarkozy A, Dallapiccola B. KBG syndrome. Orphanet J Rare Dis. 2006;1: 50.
Skjei KL, Martin MM, Slavotinek AM. KBG syndrome: report of twins, neurological characteristics, and delineation of diagnostic criteria. Am J Med Genet A. 2007;143A: 292–300.
Morghen I, Ferri E. The KBG syndrome: Case report. Cases J. 2008;1: 186.
Nicolini F, Beghi C, Gherli T. Aortic valve regurgitation in a patient affected by KBG syndrome. J Heart Valve Dis. 2009;18: 122–124.
Kumar H, Prabhu N, Cameron A. KBG syndrome: review of the literature and findings of 5 affected patients. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;108: e72–9.
Hah M, Lotspeich LJ, Phillips JM, Torres AD, Cleveland SC, Hallmayer JF. Twins with KBG syndrome and autism. J Autism Dev Disord. 2009;39: 1744–1746.
Willemsen MH, Fernandez BA, Bacino CA, Gerkes E, de Brouwer APM, Pfundt R, et al. Identification of ANKRD11 and ZNF778 as candidate genes for autism and variable cognitive impairment in the novel 16q24.3 microdeletion syndrome. Eur J Hum Genet. 2010;18: 429–435.
Oegema R, Schot R, de Wit MCY, Lequin MH, Oostenbrink R, de Coo IFM, et al. KBG syndrome associated with periventricular nodular heterotopia. Clin Dysmorphol. 2010;19: 164–165.
Almandey AH, Anthonappa RP, King NM, Fung C-W. KBG syndrome: clinical features and specific dental findings. Pediatr Dent. 2010;32: 439–444.
Youngs E, Hellings J and Butler M. ANKRD11 gene deletion in a 17 year-old-man. Clin Dysmorphol. 2011;20:170-171.
Sirmaci A, Spiliopoulos M, Brancati F, Powell E, Duman D, Abrams A, et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. 2011;89: 289–294.
Sacharow S, Li D, Fan YS, Tekin M. Familial 16q24.3 microdeletion involving ANKRD11 causes a KBG-like syndrome. Am J Med Genet A. 2012;158A: 547–552.
Lo-Castro A, Brancati F, Digilio MC, Garaci FG, Bollero P, Alfieri P, et al. Neurobehavioral phenotype observed in KBG syndrome caused by ANKRD11 mutations. Am J Med Genet B Neuropsychiatr Genet. 2013;162B: 17–23.
Khalifa M, Stein J, Grau L, Nelson V, Meck J, Aradhya S, et al. Partial deletion of ANKRD11 results in the KBG phenotype distinct from the 16q24.3 microdeletion syndrome. Am J Med Genet A. 2013;161A: 835–840.
Xu M, Zhou H, Yong J, Cong P, Li C, Yu Y, et al. A Chinese patient with KBG syndrome and a 9q31.2-33.1 microdeletion. Eur J Med Genet. 2013;56: 245–250.
Miyatake S, Murakami A, Okamoto N, Sakamoto M, Miyake N, Saitsu H, et al. A de novo deletion at 16q24.3 involving ANKRD11 in a Japanese patient with KBG syndrome. Am J Med Genet A. 2013;161A: 1073–1077.
Spengler S, Oehl-Jaschkowitz B, Begemann M, Hennes P, Zerres K, Eggermann T. Haploinsufficiency of ANKRD11 (16q24.3) Is Not Obligatorily Associated with Cognitive Impairment but Shows a Clinical Overlap with Silver-Russell Syndrome. Mol Syndromol. 2013;4: 246–249.
Tunovic S, Barkovich J, Sherr EH, Slavotinek AM. De novo ANKRD11 and KDM1A gene mutations in a male with features of KBG syndrome and Kabuki syndrome. Am J Med Genet A. 2014;164A: 1744–1749.
Lim J-H, Seo E-J, Kim Y-M, Cho H-J, Lee J-O, Cheon CK, et al. A de novo microdeletion of ANKRD11 gene in a Korean patient with KBG syndrome. Ann Lab Med. 2014;34: 390–394.
Ansari M, Poke G, Ferry Q, Williamson K, Aldridge R, Meynert AM, et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J Med Genet. 2014;51: 659–668.
Gallagher D, Voronova A, Zander MA, Cancino GI, Bramall A, Krause MP, et al. Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Dev Cell. 2015;32: 31–42.
Walz K, Cohen D, Neilsen PM, Foster J 2nd, Brancati F, Demir K, et al. Characterization of ANKRD11 mutations in humans and mice related to KBG syndrome. Hum Genet. 2015;134: 181–190.
Kim HJ, Cho E, Park JB, Im WY, Kim HJ. A Korean family with KBG syndrome identified by ANKRD11 mutation, and phenotypic comparison of ANKRD11 mutation and 16q24.3 microdeletion. Eur J Med Genet. 2015;58: 86–94.
Crippa M, Rusconi D, Castronovo C, Bestetti I, Russo S, Cereda A, et al. Familial intragenic duplication of ANKRD11 underlying three patients of KBG syndrome. Mol Cytogenet. 2015;8: 20.
Reynaert N, Ockeloen CW, Sävendahl L, Beckers D, Devriendt K, Kleefstra T, et al. Short Stature in KBG Syndrome: First Responses to Growth Hormone Treatment. Horm Res Paediatr. 2015;83: 361–364.
Hafiz A, Mufeed A, Ismael M, Alam M. An unusual case of KBG syndrome with unique oral findings. BMJ Case Rep. 2015;2015. doi:10.1136/bcr-2015-210352
Ockeloen CW, Willemsen MH, de Munnik S, van Bon BWM, de Leeuw N, Verrips A, et al. Further delineation of the KBG syndrome phenotype caused by ANKRD11 aberrations. Eur J Hum Genet. 2015;23: 1176–1185.
Samanta D, Willis E. Electroencephalographic findings in KBG syndrome: a child with novel mutation in ANKRD11 gene. Acta Neurol Belg. 2015;115: 779–782.
Parenti I, Gervasini C, Pozojevic J, Graul-Neumann L, Azzollini J, Braunholz D, et al. Broadening of cohesinopathies: exome sequencing identifies mutations in ANKRD11 in two patients with Cornelia de Lange-overlapping phenotype. Clin Genet. 2016;89: 74–81.
Low K, Ashraf T, Canham N, Clayton-Smith J, Deshpande C, Donaldson A, et al. Clinical and genetic aspects of KBG syndrome. Am J Med Genet A. 2016;170: 2835–2846.
Goldenberg A, Riccardi F, Tessier A, Pfundt R, Busa T, Cacciagli P, et al. Clinical and molecular findings in 39 patients with KBG syndrome caused by deletion or mutation of ANKRD11. Am J Med Genet A. 2016;170: 2847–2859.
Kleyner R, Malcolmson J, Tegay D, Ward K, Maughan A, Maughan G, et al. KBG syndrome involving a single-nucleotide duplication in ANKRD11. Cold Spring Harb Mol Case Stud. 2016;2: a001131.
Murray N, Burgess B, Hay R, Colley A, Rajagopalan S, McGaughran J, et al. KBG syndrome: An Australian experience. Am J Med Genet A. 2017; doi:10.1002/ajmg.a.38121
Novara F, Rinaldi B, Sisodiya SM, Coppola A, Giglio S, Stanzial F, et al. Haploinsufficiency for ANKRD11-flanking genes makes the difference between KBG and 16q24.3 microdeletion syndromes: 12 new cases. Eur J Hum Genet. 2017;25: 694–701.
Srivastava P, Gambhir PS, Phadke SR. KBG syndrome: 16q24.3 microdeletion in an Indian patient. Clin Dysmorphol. 2017;26: 161–166.
Kaname T, Yanagi K. A commentary on ANKRD11 variants cause variable clinical features associated with KBG syndrome and Coffin-Siris-like syndrome. J Hum Genet. 2017;62: 739–740.
Miyatake S, Okamoto N, Stark Z, Nabetani M, Tsurusaki Y, Nakashima M, et al. ANKRD11 variants cause variable clinical features associated with KBG syndrome and Coffin-Siris-like syndrome. J Hum Genet. 2017;62: 741–746.
Low KJ, Hills A, Williams M, Duff-Farrier C, McKee S, Smithson SF. A splice-site variant in ANKRD11 associated with classical KBG syndrome. Am J Med Genet A. 2017;173: 2844–2846.
Bianchi P. and al. Audiological findings in a de novo mutation of ANKRD11 gene in KBG syndrome: Report of a case and review of the literature. International journal of pediatric otorhinolaryngology. 2017;103:109-112
Swols DM, Foster J 2nd, Tekin M. KBG Syndrome. Orphanet journal of rare disease. 2017; 12:183
Dongen, Linde C. M. van, Ellen Wingbermühle, Wouter Oomens, Anja G. Bos-Roubos, Charlotte W. Ockeloen, Tjitske Kleefstra, and Jos I. M. Egger. 2017. “Intellectual Profiles in KBG-Syndrome: A Wechsler Based Case-Control Study.” Frontiers in Behavioral Neuroscience 11 (December):248.
association de patients américaine http://www.kbgfoundation.com/what-is-kbg-.html
Auteurs :
Valentin Ruault
Professeur David Geneviève
Professionnels de santé en Occitanie
Centre de Référence des Anomalies du Développement et Syndromes Malformatifs : pour le diagnostic et la coordination des prises en charge à tout âge de la vie.
Hôpital Arnaud de Villeneuve - CHU de Montpellier
Département de génétique médicale
34295 Montpellier cedex 5
Cardiopédiatrie - : pour le suivi des cardiopathies de l’enfant et de l’adolescent Centre de compétence
Hôpital Arnaud de Villeneuve - CHU de Montpellier
Département de pédiatrie (pôle enfant)
34295 Montpellier cedex 5
Cardiologie adulte : pour le suivi des cardiopathies à l’âge adulte Centre de compétence
Hôpital Arnaud de Villeneuve - CHU de Montpellier
Département de Cardiologie et Maladies Vasculaires
34295 Montpellier cedex 5
Chirurgie orthopédique et plastique infantile : pour les scolioses, anomalies du palais et les problèmes squelettiques Centre de compétence
Hôpital Lapeyronie
Département de chirurgie infantile (Pôle Enfant)
34295 Montpellier cedex 5
Endocrinologie pédiatrique : pour les troubles de la croissance et de la puberté Centre de compétence
Hôpital Arnaud de Villeneuve - CHU de Montpellier
Département de pédiatrie (pôle enfant)
34295 Montpellier cedex 5
Gastrologie pédiatrique : pour les difficultés alimentaires et troubles du transit Centre de compétence
Hôpital Arnaud de Villeneuve - CHU de Montpellier
Département de Pédiatrie (Pôle Enfant)
34295 Montpellier cedex 5
Néphrologie pédiatrique : pour les anomalies rénales Centre de référence
Hôpital Arnaud de Villeneuve
Département de Pédiatrie (Pôle Enfant)
34295 Montpellier cedex 5
Neuropédiatrie et neurochirurgie pédiatrique : pour le suivi développemental et des complications neurologiques Centre de compétence
Hôpital Gui de Chauliac
Département de Pédiatrie
34295 Montpellier cedex 5
Ophtalmologie pédiatrique : pour le dépistage et la surveillance des troubles de la vue Centre de référence
Hôpital Gui de Chauliac
Département d’ophtalmologie
34295 Montpellier cedex 5
ORL pédiatrique : pour le dépistage et la surveillance des troubles de l’audition Centre de référence
Hôpital Gui de Chauliac
Département ORL et chirurgie cervico-faciale (Pôle Neurosciences Tête et Cou)
34295 Montpellier cedex 5
Pédiatrie spécialisée : pour les problèmes endocriniens (taille, puberté)
Hôpital Arnaud de Villeneuve
Département de Pédiatrie (Pôle Enfant)
34295 Montpellier cedex 5
Psychiatrie de l’enfant et de l’adolescent : en cas de troubles comportementaux Centre de compétence Centre du secteur territorial en première intention
SMPEA Peyre Plantade
34295 Montpellier cedex 5
Stomatologie pédiatrique : pour la surveillance dentaire Centre de compétence
Hôpital Gui de Chauliac
Département ORL et chirurgie stomatologique (Pôle Neurosciences Tête et Cou)
34295 Montpellier cedex 5
Urologie pédiatrique : pour les problèmes des voies urinaires et cryptorchidies Centre de compétence
Hôpital Lapeyronie
Département de chirurgie infantile (Pôle Enfant)
34295 Montpellier cedex 5