Guide pratique : Syndrome de Coffin-Siris
Auteurs : Camille CENNI, interne de génétique médicale et Pr David GENEVIEVE Centre de Référence Anomalies du Développement CHU MONTPELLIER
Publié mars 2020
OMIM #135900 ; ORPHA :1465)
Qu'est-ce que le syndrome de Coffin-Siris (SCS) ?
Le syndrome de Coffin-Siris (SCS) est une maladie génétique pouvant concerner plusieurs organes, congénitale (c’est-à-dire apparue pendant la vie fœtale), décrite pour la première fois en 1970 par Grange Coffin et Evelyn Siris.
Il est classiquement caractérisé par :
- une aplasie ou une hypoplasie de la phalange distale ou de l'ongle du cinquième doigt,
- un retard de développement ou un déficit intellectuel,
- une hypotonie,
- des particularités morphologiques du visage,
- un hirsutisme principalement au niveau du visage, des bras et du dos, associé à des cheveux clairsemés.
Le syndrome de Coffin-Siris était initialement considéré comme une maladie très rare, car la prévalence à la naissance était estimée inférieure à 1/1 000 000.
Aspects génétiques du syndrome de Coffin-Siris
Actuellement, le gène majoritairement impliqué dans ce syndrome est l’un des gènes le plus fréquent rapporté dans la déficience intellectuelle.
Il est également, très hétérogène d’un point de vu génétique, puisqu’actuellement 11 gènes peuvent être responsables de ce syndrome (cf Diagnostic génétique et moléculaire).
Le diagnostic génétique est important à poser afin de mettre en place le plus tôt possible les aides nécessaires au développement optimal des enfants et de prévenir les éventuelles complications liées aux atteintes de certains organes, comme le cœur par exemple.
Le diagnostic génétique du syndrome de Coffin-Siris repose sur l’identification d’une anomalie génétique dans l’un des gènes impliqués dans ce syndrome.
La confirmation moléculaire est importante pour le conseil génétique à donner aux parents et aux personnes à l’âge adulte.
|
Pathologie |
Gène |
Num OMIM de la maladie |
Num OMIM du gène |
|
SCS1 |
ARID1B |
# 135900 |
*614556 |
|
SCS2 |
ARID1A |
# 614607 |
*603024 |
|
SCS3 |
SMARCB1 |
# 614608 |
*601607 |
|
SCS4 |
SMARCA4 |
# 614609 |
*603254 |
|
SCS5 |
SMARCE1 |
# 616938 |
*603111 |
|
SCS6 |
ARID2 |
# 617808 |
*609539 |
|
SCS7 |
DPF2 |
# 618027 |
*601671 |
|
SCS8 |
SMARCC2 |
# 618362 |
*601734 |
|
SCS9 |
SOX11 |
# 615866 |
*600898 |
|
SCS10 |
SOX4 |
# 618506 |
*184430 |
|
SCS11 |
SMARCD1 |
# 618779 |
*601735 |
Corrélations Génotype-Phénotype
Les tableaux ci-dessous reprennent la description du syndrome en fonction du gène impliqué.
Tableau de comparaison génotypes-phénotypes français
Tableau de comparaison génotypes-phénotypes anglais
Fréquence des gènes impliqués dans le syndrome de Coffin-Siris, selon une revue de la littérature (mise à jour, mars 2020).

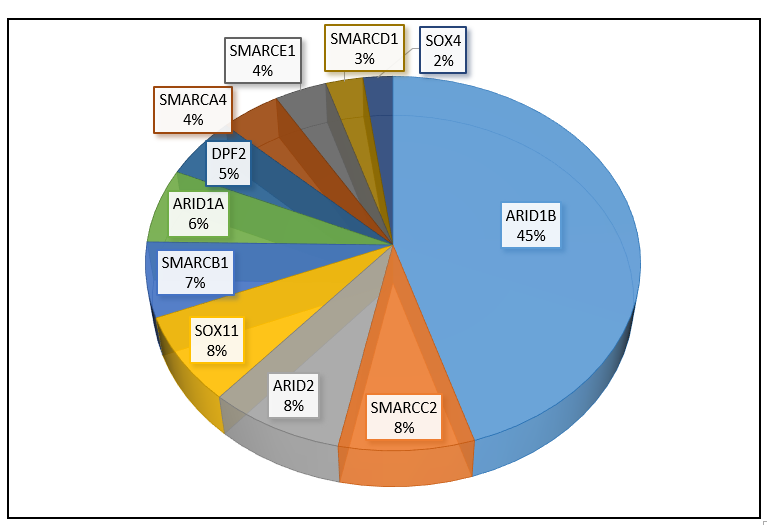

Dans 25% des situations, aucune anomalie génétique n’est identifiée. Cela ne signifie pas qu’il ne s’agit pas d’un syndrome de Coffin-Siris, mais que la cause génétique est encore inconnue (anomalie présente dans un des 11 gènes connus, mais non identifiée par les techniques actuelles ou présente dans un gène non encore décrit dans cette pathologie).
Caractéristiques cliniques du syndrome
Le syndrome de Coffin-Siris est un ensemble de malformations congénitales regroupant un large éventail de symptômes.
Les signes pendant la grossesse sont rares (bébé de petite taille et/ou avec une petite tête, anomalies cardiaques ou du système nerveux central).
Certains signes, notamment les anomalies malformatives, peuvent être notés dès la période néonatale :
- une hypoplasie ou une aplasie de la phalange distale/ongle du cinquième doigt, c’est-à-dire une phalange ou un ongle de plus petite taille ou absent (chez pratiquement tous les enfants),
- des difficultés alimentaires ayant un impact sur la croissance (dans 90% des situations),
- une hypotonie (dans 75% des situations),
- une épilepsie (dans 50% des situations),
- des problèmes visuels tels qu’un strabisme (environ 50% des situations), un ptosis (c’est-à-dire une paupière tombante, environ 50% des situations) ou une myopie (20% des situations),
- des problèmes auditifs souvent associés à des infections ORL (dans 45% des situations),
- des anomalies cardiaques (dans 35% des situations) telles qu’une communication interventriculaire ou interauriculaire, une tétralogie de Fallot ou une persistance du canal artériel,
- des malformations génito-urinaires et rénales (environ 35% des situations) telles qu’une cryptorchidie (un défaut de descente des testicules dans les bourses) ou des reins en fer à cheval,
- des anomalies du système nerveux central, tel qu’une malformation de Dandy-Walker, une simplification de la gyration cérébrale ou une agénésie du corps calleux.
Les particularités morphologiques peuvent être présentes dès la naissance dans environ 30% des situations, notamment l’hypertrichose (c’est-à-dire une pilosité trop importante) ou la microcéphalie (c’est-à-dire une petite tête).
Dans l’enfance, il est décrit :
- un retard de développement ou cognitif léger à sévère (chez tous les patients). En moyenne, les enfants atteints du syndrome de Coffin-Siris se tiennent assis vers 12 mois, marchent vers 30 mois et disent leur premier mot vers 24 mois. L’expression orale est plus sévèrement affectée que la compréhension.
- des particularités morphologiques du visage, chez tous les patients. Ces caractéristiques s’intensifient généralement avec le temps.
- une hypertrichose, principalement au niveau du dos, des bras et des épaules (95% des situations),
- des anomalies musculo-squelettiques tel qu’une clinodactylie (une légère courbure d’un doigt dans le plan de la main, dans environ 40% des situations), un âge osseux, typiquement retardé de 2 à 3 ans (environ 40% des situations), une laxité ligamentaire (environ 65% des situations), une scoliose (environ 30% des situations) ou des hernies (environ 10% des situations),
- des cheveux clairsemés et fins précocement (environ 60% des situations),
- des infections ORL virales plus fréquentes (environ 60% des situations),
- des difficultés alimentaires persistant dans l’enfance entrainant un retard de croissance. Le poids et la taille sont inferieurs au 5ieme percentile dans environ 20% des situations.
- des troubles du comportement tels qu’une hyperactivité, une agressivité ou des traits autistiques peuvent être présents (environ 10% des situations).
Diagnostic
Le diagnostic du syndrome de Coffin-Siris repose soit sur l’identification de signes cliniques compatibles associés à une anomalie génétique dans l’un des gènes impliqués dans ce syndrome, soit lors d’une analyse pangénomique sans idée préconçue.
On considère qu’il existe des signes majeurs et des signes mineurs de la maladie.
Les signes caractéristiques majeurs comprennent :
- le retard de développement,
- l’hypoplasie ou une aplasie de la phalange distale/ongle du cinquième doigt,
- les caractéristiques morphologiques du visage.
Les caractéristiques mineures incluent :
- le retard de croissance staturo-pondérale,
- les difficultés d’alimentation,
- la microcéphalie,
- les manifestations ophtalmologiques,
- les anomalies cardiaques,
- l’hypertrichose et les cheveux clairsemés,
- l’atteinte neurologique,
- l’atteinte auditive,
- la laxité ligamentaire,
- les malformations génito-urinaires et rénales
et les infections fréquentes.
Conseil génétique
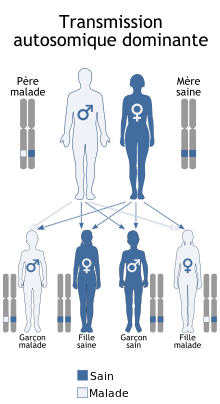
Le syndrome de Coffin-Siris est une des maladies génétiques les plus fréquentes dans la déficience intellectuelle, de transmission dite « autosomique dominante ».
Nous avons tous nos gènes en deux exemplaires, un venant de notre mère et l’autre venant de notre père.
Le terme « autosomique » signifie que les personnes atteintes peuvent aussi bien être des femmes que des hommes, la maladie n’est pas liée au sexe. Le terme « dominant » signifie qu’il suffit qu’une seule copie d’un des gènes impliqués dans ce syndrome soit altérée pour développer la maladie. Il s’agit le plus fréquemment d’une anomalie génétique « de novo ». Cela signifie que ni la mère, ni le père ne sont porteurs de cette anomalie et qu’elle est apparue accidentellement, probablement lors de la formation de l’ovocyte ou du spermatozoïde qui ont permis la naissance de l’enfant.
Cela implique pour les parents que le risque d’avoir un deuxième enfant atteint de ce syndrome est très faible. Cependant, ce risque n’étant jamais égal à zéro, il peut être proposé aux parents un diagnostic prénatal invasif ou non-invasif pendant la grossesse, si l’anomalie familiale est connue.
Pour les personnes atteintes du syndrome de Coffin-Siris, lorsqu’elles souhaitent avoir des enfants, elles ont 50% de risques de leur transmettre l’anomalie, quel que soit le sexe, et donc 50% de chances de ne pas leur transmettre.
Prise en charge médicale initiale
Au moment du diagnostic, l’évaluation initiale cherche à faire le point sur l’étendue de la maladie et les problèmes médicaux à prendre en charge. Elle est le plus souvent coordonnée par un généticien clinicien ou un pédiatre du Centre de Référence « Anomalies du Développement et Syndromes Malformatifs » en relation avec le pédiatre ou le médecin traitant de l’enfant. Selon les complications associées, d’autres spécialistes pourront intervenir dans le suivi.
Bilan clinique
- Consultation avec un généticien clinicien pour une première évaluation, pose du diagnostic, réalisation de l’étude génétique et coordination avec les autres intervenants.
- Consultation avec un neurologiste pédiatre afin de faire le point sur les difficultés neurologiques et les aides à mettre en place.
- Évaluations kinésithérapique, ergologique, orthophonique et psychométrique, en fonction de l’âge.
- Consultation avec un gastro-pédiatre et un nutritionniste afin d’évaluer les difficultés alimentaires et de mettre en place des mesures permettant une prise de poids la plus optimale possible.
- Consultation ophtalmologique avec évaluation du fond d’œil et mesure de l’acuité visuelle (en fonction de l’âge de l’enfant).
Bilan complémentaire systématique
- Fond d’œil et mesure de l’acuité visuelle.
- Évaluation audiologique appropriée en fonction de l’âge.
- Échographie cardiaque à la recherche d’une anomalie morphologique.
- Échographie rénale et génito-urinaire à la recherche d’une anomalie morphologique.
- IRM cérébrale à la recherche d’une anomalie morphologique (malformation de Dandy-Walker, simplification de la gyration cérébrale, agénésie du corps calleux).
Bilan complémentaire spécialisé dirigé par la clinique
- Électro-encéphalogramme à la recherche d’une épilepsie si présence de signes cliniques.
- Radiographie de la main gauche pour évaluation de l’âge osseux si nécessaire.
- Radiographie du dos si suspicion de scoliose.
Accès aux droits
- 100% ALD hors liste
- Dossier MDPH
Scolarisation
- Orientation CAMSP si nécessaire et si enfant âgé de moins de 6 ans
- PAI et/ou PPS si en âge scolaire
- Soutien scolaire si nécessaire
- Discuter de l'intérêt d’une AVS
- En fonction de l’âge, discuter l'intérêt d’une notification IME avec l’équipe d’enseignants référents et les parents
Suivi
Suivi annuel par généticien ou pédiatre pour évaluation pédiatrique classique, gastro pédiatre et nutritionniste pour suivi de la croissance, ophtalmologiste et ORL.
0-5 ans
Le diagnostic peut être porté dans la première année de vie. L’enfant bénéficiera du suivi habituel proposé par les pédiatres français.
Ce suivi sera d’autant plus nécessaire que vont apparaitre les symptômes non spécifiques comme des troubles de l’alimentation ou une hypotonie. Le bilan initial doit être réalisé dès l’évocation du diagnostic. Puis un suivi régulier par un pédiatre doit être proposé. Pour rappel, neuf consultations sont prévues au cours de la première année de vie, prises en charge à 100 % par l’Assurance Maladie, à 1, 2, 3, 4, 5, 6, 9 mois et 1 an ; puis trois consultations sont prévues au cours de la deuxième année puis deux fois par an jusqu’à six ans.
- Croissance staturo-pondérale,
- Digestif : recherche de reflux gastro-œsophagien, de trouble du transit (constipation),
- Examen neurologique à la recherche d’une hypotonie, d’anomalies de la motricité, de signes précoces de déficit intellectuel et d’une épilepsie,
- Examen cardiaque pédiatrique si existence d’une malformation cardiaque,
- Examen musculo-squelettique : hyperlaxité ligamentaire, scoliose,
- Développement psychomoteur : la marche est acquise vers l’âge de 2 ans ½. Les compétences linguistiques de compréhension sont toujours bien meilleures que la compétence en expression orale. Une prise en charge avec des moyens adaptés en communication non verbale doit être rapidement mise en place afin de ne pas voir apparaitre des troubles des comportements surajoutés. Il importe de s’assurer que la famille est en lien avec les équipes de prise en charge médico-éducatives, sociales et psychologiques (CAMSP ou autres, MDPH…),
- Examen ophtalmologique. Les troubles sont peu spécifiques, strabisme en premier, mais myopie, ptosis sont aussi retrouvés.
- Examen ORL (surveillance régulière de l’audition, notamment en cas d’otites à répétition),
- Suivi urogénital et rénal en cas de malformation,
- Les troubles du comportement sont possibles : hyperactivité, agressivité, trait autistique. Il est important de ne pas sur-traiter médicalement ce trouble du comportement.
- Vaccinations habituelles, à poursuivre aux âges habituels.
5-12 ans
- Croissance staturo-pondérale,
- Digestif : recherche de reflux gastro-œsophagien, de trouble du transit (constipation),
- Examen neurologique. La recherche d’épilepsie doit être poursuivie et sa prise en charge continue, les crises pouvant être inapparentes pour la famille,
- Examen cardiaque pédiatrique si existence d’une malformation cardiaque,
- Il importe de s’assurer que la famille est en lien avec les équipes de prise en charge médico-éducatives, sociales et psychologiques (IME / IMPRO, SESSAD, ou autres, MDPH…),
- Le suivi ophtalmologique (ophtalmologue et orthoptiste) est poursuivi et les traitements adaptés,
- Examen ORL (surveillance régulière de l’audition, notamment en cas d’otites à répétition),
- Suivi urogénital et rénal en cas de malformation,
- Poursuite des vaccinations habituelles.
12-18 ans
A cet âge les adolescents ont en général une bonne santé. L’âge de la puberté et son déroulement ne posent pas de problème particulier.
- Clinique : Un examen clinique complet annuel avec surveillance des pathologies de l’enfant.
- Examen neurologique.
- Examen cardiaque.
- Examen ophtalmologique.
- Examen ORL.
- Dépistage des problèmes orthopédiques (rachis tout particulièrement avec possibilité de scoliose, à surveiller tous les 6 mois lors de la croissance pubertaire).
Il importe de rechercher tôt un établissement de prise en charge à l’âge adulte.
Adultes
La surveillance médicale doit se poursuivre en fonction des pathologies notées pendant l’enfance.
- Une attention particulière sera portée sur le suivi cardiaque, neurologique, moteur, sensoriel, orthopédique, dentaire et sur les troubles psychologiques ou psychiatriques.
- Les adultes vivent souvent dans des foyers spécialisés : Foyers d’Accueil Médicalisés (FAM) ou dans des Maisons d’Accueil Spécialisé (MAS). L’espérance de vie est apparemment non diminuée.
- Les dépistages systématiques des cancers de l’adulte (sein, colon) proposés en population générale doivent aussi faire partie de la prise en charge selon les modalités habituelles.
Suivi paramédical
- La kinésithérapie
La prise en charge débute souvent avant que le diagnostic ait été fait. Elle commence donc dès la constatation des difficultés motrices. Elle vise à faciliter le tonus avec comme objectif l’acquisition de la marche et d’améliorer celle-ci. Elle doit commencer très tôt, dès les premiers mois de vie. Sa poursuite permet outre d’acquérir la marche, d’en améliorer sa qualité et de lutter contre les troubles de l’équilibre. À l’âge adulte, la kinésithérapie entretient l’acquisition de la marche et l’autonomie.
- L’orthophonie
Une prise en charge orthophonique précoce est nécessaire dès la petite enfance (en cas de trouble de succion/déglutition) pour mettre en place une stimulation oro-faciale. Elle est fondamentale pour assurer un moyen de communication. Les séances d’orthophonie doivent débuter très tôt pour corriger ces problèmes d’hypotonie de la sphère linguale et pour diminuer la salivation excessive puis pour stimuler et aider l’apprentissage du langage oral.
- La psychomotricité
Elle vise à améliorer les troubles de la motricité fine. Elle permet d’améliorer la coordination des mouvements et la précision des gestes, surtout si une ataxie ou des tremblements sont présents. L’enfant utilisera plus facilement ses mains pour attraper et manipuler les objets.
- L’ergothérapie
Elle est utile en cas de trouble grapho-moteur à l’école. Elle peut également aider les personnes au mieux dans la réalisation des tâches quotidiennes en tenant compte de l’environnement. L’objectif de l’ergothérapie est de permettre à l’individu d’acquérir une meilleure autonomie individuelle, sociale et professionnelle en tenant compte de l’environnement.
Association de malades
Groupe Facebook : « Syndrome de Coffin-Siris »
Références et sites internet
OMIM :
Type 1 : https://omim.org/entry/135900?search=coffin%20siris&highlight=coffin%20siri
Type 2 : https://omim.org/entry/614607?search=coffin%20siris&highlight=coffin%20siri
Type 3 : https://omim.org/entry/614608?search=coffin%20siris&highlight=coffin%20siri
Type 4 : https://omim.org/entry/614609?search=coffin%20siris&highlight=coffin%20siri
Type 5 : https://omim.org/entry/616938?search=coffin%20siris&highlight=coffin%20siri
Type 6 : https://omim.org/entry/617808?search=coffin%20siris&highlight=coffin%20siri
Type 7 : https://omim.org/entry/618027?search=coffin%20siris&highlight=coffin%20siri
Type 8 : https://omim.org/entry/618362?search=coffin%20siris&highlight=coffin%20siri
Type 9 : https://omim.org/entry/615866?search=coffin%20siris&highlight=coffin%20siri
Type 10 :
https://omim.org/entry/618506?search=coffin%20siris%2010&highlight=10%20coffin%20siri
Type 11 : https://omim.org/entry/618779?search=coffin%20siris&highlight=coffin%20siri
GeneReviews : https://www.ncbi.nlm.nih.gov/books/NBK131811/
Professionnels de santé en Occitanie Toute anomalie mise en évidence lors du bilan initial et lors du suivi doit être prise en charge par l’équipe médicale spécialisée.
Centre de Référence des Anomalies du Développement et Syndromes Malformatifs : pour le diagnostic et la coordination des prises en charge à tout âge de la vie.
Hôpital Arnaud de Villeneuve - CHRU Montpellier
Pôle Biologie Pathologie - Département de Génétique Médicale
34295 Montpellier cedex 5
Neurologie pédiatrique : pour la prise en charge des complications neurologiques chez l’enfant
Hôpital Gui de Chauliac - CHRU Montpellier
Pôle Femme Mère Enfant - Département de Neuropédiatrie
34295 Montpellier cedex 5
Neurologie adulte : pour la prise en charge des complications neurologiques chez l’adulte
Hôpital Gui de Chauliac - CHRU Montpellier
Pôle Neuroscience Tête et Cou - Département de Neurologie
34295 Montpellier cedex 5
Cardiologie pédiatrique : pour la prise en charge des anomalies cardiaques
Hôpital Arnaud de Villeneuve - CHRU Montpellier
Pôle Femme Mère Enfant - Département de Pédiatrie
34295 Montpellier cedex 5
Cardiologie adulte : pour le suivi à l’âge adulte
Hôpital Arnaud de Villeneuve - CHRU Montpellier
Pôle Cœur Poumons - Département de Cardiologie et Maladie Vasculaire
34295 Montpellier cedex 5
Hépato-Gastro-Entérologie pédiatrique : pour la prise en charge des complications digestives et nutritionnelles
Hôpital Arnaud de Villeneuve - CHRU Montpellier
Pôle Femme Mère Enfant - Département de Pédiatrie
34295 Montpellier cedex 5
Gastrologie adulte : pour le suivi à l’âge adulte
Hôpital Saint-Eloi - CHRU Montpellier
Pôle Digestif - Département d’Hépato-gastro-entérologie
34295 Montpellier cedex 5
Néphrologie pédiatrique et adulte : pour le dépistage et le suivi
Hôpital Lapeyronie - CHRU Montpellier
Pôle EMMBRUN - Département Néphrologie
34295 Montpellier cedex 5
ORL pédiatrique et adulte : pour le dépistage, la surveillance et le suivi des troubles de l’audition
Hôpital Gui de Chauliac - CHRU Montpellier
Pôle Neurosciences Tête et Cou - Département d’ORL et de Chirurgie cervico-faciale
34295 Montpellier cedex 5
Orthopédie pédiatrique : pour la prise en charge des complications orthopédiques, notamment la scoliose
Hôpital Lapeyronie - CHRU Montpellier
Pôle Femme Mère Enfant - Département de Chirurgie Infantile
34295 Montpellier cedex 5
Orthopédie adulte : pour le suivi à l’âge adulte
Hôpital Lapeyronie - CHRU Montpellier
Pôle Os et Articulations - Département de Chirurgie orthopédique et de Traumatologie
34295 Montpellier cedex 5
Ophtalmologie pédiatrique et adulte : pour le dépistage et le suivi des troubles visuels
Hôpital Gui de Chauliac - CHRU Montpellier
Pôle Neurosciences Tête et Cou - Département Ophtalmologie
34295 Montpellier cedex 5