Guide pratique : Syndrome de Sotos
Auteurs : Pauline MONIN, interne de génétique médicale et Pr David GENEVIEVE Centre de Référence Anomalies du Développement CHU MONTPELLIER
Publié avril 2020
OMIM : #117550
Orphanet : ORPHA 821
Qu'est-ce que le syndrome de Sotos ?
Le
Le nombre de personnes touchées par ce syndrome n’est pas connu de façon précise mais des centaines ont été décrites dans la littérature médicale. La fréquence du syndrome de Sotos est estimée à environ 1 personne sur 14000.
La prise en charge de cette pathologie est multidisciplinaire et nécessite un suivi régulier spécialisé, à intervalles plus ou moins rapprochés selon la sévérité des symptômes.
Caractéristiques cliniques du syndrome
Les symptômes observés dans le syndrome de Sotos sont classés en 3 groupes selon la fréquence à laquelle ils sont retrouvés chez les personnes décrites dans la littérature médicale.
Premièrement, on distingue les symptômes dits « cardinaux », présents chez plus de 90% des personnes porteuses du syndrome du Sotos. Cette catégorie comprend :
- Une avance de croissance en taille et/ou du périmètre crânien (on parle de « macrocéphalie »). Ces particularités peuvent parfois être repérées aux échographies anténatales.
- Un décalage des acquisitions motrices et des difficultés d’apprentissage, plus ou moins sévères d’une personne à l’autre, et caractérisées par des difficultés de raisonnement non verbal et de raisonnement quantitatif. Les personnes ont plus de facilité sur le plan verbal, ainsi qu’une bonne mémoire visuo-spatiale.
- Des particularités morphologiques du visage, avec, en particulier, un front pouvant être décrit comme « large » et proéminent. Le visage est plutôt allongé et étroit dans l’enfance. L’implantation des cheveux peut être éparse au niveau frontal. Ces particularités ont tendance à s’atténuer à l’âge adulte.
L’association de ces 3 symptômes fait fortement suspecter le diagnostic de syndrome de Sotos.
Deuxièmement, on distingue les symptômes dits « majeurs », présents chez 15 à 89% des personnes porteuses du syndrome de Sotos. Cette catégorie comprend :
- Des troubles du comportement, et en particulier les troubles du spectre autistique, les phobies et une possible agressivité (automutilation et hétéro-agressivité). Environ 83% des personnes présentent des signes en faveur d’un trouble du spectre autistique, et ces signes sont particulièrement observés entre l’âge de 5 ans et l’âge de 19 ans.
- Une avance d’âge osseux (diagnostiquée par une radiographie de la main et du poignet). 75 à 80% des enfants pré-pubères porteurs d’un syndrome de Sotos sont concernés.
- Des complications néonatales, souvent transitoires, telles qu’une hypotonie (environ 75% des nouveau-nés), des difficultés de succion (environ 70% des nouveau-nés), et un ictère (« jaunisse ») (environ 65% des nouveau-nés).
- Une scoliose, décrite chez environ 30% des personnes porteuses d’un syndrome de Sotos, et ne nécessitant que rarement un recours au port du corset et à la chirurgie.
- Des anomalies de la morphologie cérébrale, très variables, vues sur une IRM ou un scanner cérébral(e). La plus fréquente est la dilatation des ventricules cérébraux (ou « hydrocéphalie »), et concerne environ 28% des personnes.
- Une épilepsie : environ 25% des personnes décrites ont présenté une ou plusieurs crises épileptiques au cours de leur vie. Différents types de crises ont été décrits.
- Des anomalies cardiaques, décrites chez environ 20% des personnes, plus ou moins sévères et complexes selon les situations. Cela peut concerner les enfants comme les adultes.
- Une
hyperlaxité articulaire , avec ou sans pieds plats. Ce symptôme est décrit chez au moins 20% des personnes. - Des anomalies rénales, qui concernent environ 15% des personnes porteuses d’un syndrome de Sotos. La plus fréquente est le reflux vésico-urétéral.
- Et une
pré-éclampsie maternelle pendant la grossesse. Il s’agit d’une hypertension artérielle maternelle, pouvant se compliquer sévèrement en l’absence de traitement. Cela concerne environ 15% des grossesses d’enfants porteurs d’un syndrome de Sotos.
Enfin, il existe des symptômes dits « associés », présents chez 2 à 15% des personnes porteuses du syndrome de Sotos, et dont le plus commun est la constipation, rentrant parfois dans le cadre d’une maladie de Hirschprung. La maladie de Hirschprung est une « paralysie » intestinale liée à l’absence de ganglions nerveux dans la paroi du gros intestin. Ce défaut d’innervation se traduit par un arrêt des contractions intestinales permettant la progression des selles dans le tube digestif (arrêt du « péristaltisme »). Ceci a comme conséquence un arrêt de la progression des selles, associé à une dilatation de l’intestin en amont de sa partie malade dénervée, et une constipation.
Un reflux gastro-œsophagien peut également être présent dans le syndrome de Sotos.
Une surdité de transmission est possible, plus fréquemment que dans la population générale.
Et il existe un sur-risque de développer des tumeurs, notamment des tumeurs malignes (cancers). Cela concerne environ 3% des personnes porteuses d’un syndrome de Sotos, quel que soit l’âge.
Le diagnostic de
Cette suspicion doit ensuite être confirmée par la réalisation d’analyses génétiques.
Aspects génétiques du syndrome de Sotos
Le gène en cause dans le syndrome de Sotos est le gène NSD1 (également appelé KMT3B). Ce gène code pour une protéine qui régule l’expression d’autres gènes (protéine de type lysine-histone-méthyltransférase). Le syndrome de Sotos fait donc partie des chromatinopathies.
Le gène NSD1 est situé sur le bras long chromosome 5 (dans la région 5q35 du chromosome 5).
Nous possédons tous 46 chromosomes au sein de nos cellules. Ces 46 chromosomes sont répartis en 23 paires avec, pour chaque paire, un chromosome hérité de notre mère et un chromosome hérité de notre père. Parmi ces 23 paires de chromosomes, on distingue les autosomes (chromosomes 1 à 22) qui sont communs aux filles et aux garçons, et la paire de gonosomes qui sont les chromosomes sexuels : XX chez les filles et XY chez les garçons.
Le syndrome de Sotos est une pathologie dite « autosomique dominante » ce qui signifie qu’elle est liée à un autosome (chromosome 5 ici), et que l’atteinte d’une seule des 2 copies du gène NSD1 est suffisante pour causer la pathologie. On parle également « d’hétérozygotie ».
Cette atteinte du gène NSD1 correspond soit à la présence d’un variant pathogène ponctuel (auparavant appelé « mutation ponctuelle ») dans la séquence du gène, soit à une délétion (perte) d’une portion du chromosome 5 (délétion 5q35), emportant tout ou partie du gène. Deux techniques peuvent donc être réalisées pour aboutir au diagnostic : le séquençage génique d’une part, et l’analyse chromosomique sur puce à ADN d’autre part. Pour cela, une simple prise de sang est réalisée, qui ne nécessite pas d’être à jeun.
Corrélations génotype-phénotype : différences de symptômes entre les variants pathogènes ponctuels et les délétions
A noter qu’en cas de délétion du gène NSD1, l’avance de croissance en taille est moins importante qu’en cas de variant pathogène ponctuel.
Concernant le retard de développement et les difficultés d’apprentissage, ils sont plus sévères en cas de délétion qu’en cas de variant pathogène ponctuel.

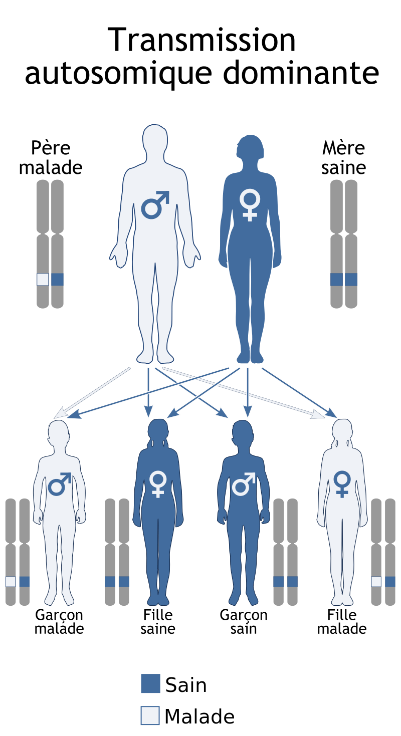


Le syndrome de Sotos est une pathologie à « pénétrance complète », ce qui signifie que tout individu porteur d’une anomalie du gène NSD1 présentera des symptômes, il n’existe pas de porteurs sains. Néanmoins, le syndrome de Sotos peut s’exprimer différemment d’un individu à l’autre : on parle « d’expressivité variable », avec parfois très peu de symptômes visibles.
Conseil génétique
Dans la majorité des situations (environ 95%), l’anomalie du gène NSD1 survient de novo, c’est-à-dire que les parents du sujet atteint ne sont pas porteurs de l’anomalie génétique identifiée chez leur enfant. Ceci nécessite d’être confirmé par la réalisation d’une prise de sang chez les deux parents. Néanmoins, cela n’exclut pas la possibilité d’un mosaïcisme germinal, c’est-à-dire que seule une petite portion des ovocytes ou des spermatozoïdes soient porteurs de l’anomalie génétique, et que celle-ci soit donc à risque d’être de nouveau transmise à la descendance (environ 1%). Toutefois, le risque de mosaïcisme germinal est rare.
Dans environ 5% des situations, l’anomalie du gène NSD1 est héritée d’un des 2 parents qui est lui-même atteint. Dans ce cas, le couple à un risque de 50% d’avoir à nouveau un enfant atteint du syndrome de Sotos.
Chaque individu porteur du syndrome a lui-même 50% de risque de le transmettre à sa descendance, et ce à chaque nouvelle grossesse. Ce risque est le même pour les individus de sexe masculin et les individus de sexe féminin.
Prise en charge médicale initiale et suivi
Lorsque le diagnostic de syndrome de Sotos est confirmé, plusieurs évaluations doivent être réalisées si cela n’a pas été fait dans le cadre de la démarche diagnostique (cf tableau ci-dessous).
De plus, bien qu’il n‘existe pas à ce jour de recommandations de suivi, plusieurs éléments doivent être surveillés chez les personnes porteuses du syndrome de Sotos (cf tableau ci-dessous).
Le suivi doit être assuré, dans le meilleur des cas, par une équipe multidisciplinaire spécialisée, au sein d’un centre de référence maladies rares (CRMR) spécialisé dans les anomalies du développement.
La fréquence du suivi est variable selon la sévérité des symptômes.
|
|
Appareil cardio-vasculaire |
Appareil génito-urinaire |
Système neurologique |
Appareil orthopédique |
Organes sensoriels (ORL / Ophtalmologie) |
Développement psychomoteur |
|
Bilan au moment du diagnostic génétique |
Auscultation cardiaque
Mesure de la pression artérielle
Consultation auprès d’un cardiologue et écho-cardiographie
|
Echographie rénale recherchant notamment un reflux vésico-urétéral
|
Recherche de signes évocateurs d’épilepsie
IRM cérébrale en cas de signe d’hyper-tension intra-crânienne
|
Examen attentif du dos à la recherche d’une scoliose
|
Bilan auditif |
Evaluation clinique, recherche de difficultés d’apprentissage |
|
Suivi |
Mesure de la pression artérielle à chaque consultation
Contrôle écho-cardiographique à l’appréciation du cardiologue
|
Examen Cyto-Bactériologique des Urines (ECBU) en cas de signes d’infection |
Recherche de signes évocateurs d’épilepsie
IRM cérébrale en cas de signe d’hyper-tension intra-crânienne
|
Examen du dos à chaque consultation
|
Examen ORL et bilan auditif selon l’évolution
Examen ophtalmologique si besoin
|
Suivi régulier par un pédiatre spécialisé et/ou un généticien |
Fiche de suivi médical à télécharger
Le risque de tumeur et cancer ne justifie pas la mise en place d’un suivi spécifique. Ainsi, une surveillance de routine n’est pas recommandée. Néanmoins, il est important que les professionnels intervenant dans la prise en charge soient sensibilisés vis-à-vis de ce risque.
Traitement
Il n’existe pas de traitement étiologique à ce jour, et l’approche thérapeutique est principalement symptomatique.
Les symptômes sont traités selon les recommandations de chaque spécialiste d’organe.
Il existe par contre des mesures de prévention. En particulier, un traitement antibiotique doit être instauré en cas de reflux vésico-urétéral, en prévention des infections urinaires.
A noter également qu’une dilatation des ventricules cérébraux (vue à l’IRM) ne nécessite pas d’intervention chirurgicale en l’absence de signe d’hypertension intracrânienne.
Scolarisation
La prise en charge du retard de développement et des difficultés d’apprentissage doit être la plus précoce possible. Il est important de favoriser une scolarisation en milieu ordinaire, avec, si besoin, mise en place d’aménagements, d’aides matérielles et d’aides humaines (Auxiliaire de Vie Scolaire, AVS). Un Projet Personnalisé de Scolarisation (PPS) peut être rédigé, en fonction des besoins, en lien avec les équipes éducatives. Dans certains cas, une orientation en milieu éducatif spécialisé est nécessaire.
Suivi paramédical
Des prises en charge rééducatives adaptées aux besoins de chaque personne doivent aussi être mises en place le plus précocement possible, telles que l’orthophonie, la psychomotricité, la kinésithérapie, le suivi par une psychologue, ... Les bilans réalisés par ces différents professionnels, et en particulier l’évaluation neuropsychologique, sont très importants puisqu’ils permettent d’identifier les points forts et les points faibles de chaque personne. Ainsi, les apprentissages scolaires seront facilités en s’appuyant sur les points forts et en compensant ou en contournant les difficultés liées aux faiblesses.
Accès aux droits
Des aides financières, telle que l’Allocation pour l’Education des Enfants en situation de Handicap (AEEH), peuvent également être mises en place pour compenser la prise en charge du handicap. L’AEEH est basée sur les constatations des professionnels médicaux et paramédicaux d’une part, et des aidants principaux d’autre part.
Le médecin référent se charge de remplir la demande de prise en charge à 100% au titre d’une Affection Longue Durée (ALD) hors liste. Il remplit également le certificat médical pour le dossier de la Maison Départementale de l’Autonomie (MDA) (anciennement appelée Maison Départementale pour les Personnes en situation de Handicap, MDPH).
Enfin, le réseau Maladies Rares Méditerranée est également à votre disposition pour vous accompagner dans les différentes démarches sociales : aide à la constitution du dossier pour la Maison Départementale de l’Autonomie (MDA), …
Association de malades
Il existe une association de personnes françaises pour le syndrome de Sotos et les troubles apparentés : il s’agit de l’association Sotos éveil qui est très active et qui propose notamment des rassemblements annuels. De nombreuses informations sont disponibles sur leur site : http://sotoseveil.fr/ .
Références et sites internet
Genereviews : https://www.ncbi.nlm.nih.gov/books/NBK1479/
OMIM : https://www.omim.org/entry/117550?search=sotos&highlight=soto
Orphanet : https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=FR&data_id=588&Disease_Disease_Search_diseaseGroup=sotos&Disease_Disease_Search_diseaseType=Pat&Maladie(s)/groupes de maladies=Syndrome-de-Sotos&title=Syndrome de Sotos&search=Disease_Search_Simple
de Boer L, Röder I, Wit JM. 2006. Psychosocial, cognitive, and motor functioning in patients with suspected
Sotos syndrome: a comparison between patients with and without NSD1 gene alterations. Dev. Med. Child Neurol. 48: 582–588.
Foster A, Zachariou A, Loveday C, Ashraf T, Blair E, Clayton-Smith J, Dorkins H, Fryer A, Gener B, Goudie D,
Henderson A, Irving M, Joss S, Keeley V, Lahiri N, Lynch SA, Mansour S, McCann E, Morton J, Motton N, Murray A, Riches K, Shears D, Stark Z, Thompson E, Vogt J, Wright M, Cole T, Tatton-Brown K. 2019. The phenotype of Sotos syndrome in adulthood: A review of 44 individuals. Am. J. Med. Genet. C Semin. Med. Genet. 181: 502–508.
Lane C, Milne E, Freeth M. 2017. Characteristics of Autism Spectrum Disorder in Sotos Syndrome. J. Autism
Dev. Disord. 47: 135–143.
Lane C, Milne E, Freeth M. 2019. The cognitive profile of Sotos syndrome. J. Neuropsychol. 13: 240–252.
Sheth K, Moss J, Hyland S, Stinton C, Cole T, Oliver C. 2015. The behavioral characteristics of Sotos
syndrome. Am. J. Med. Genet. A. 167A: 2945–2956.
Tatton-Brown K, Douglas J, Coleman K, Baujat G, Cole TRP, Das S, Horn D, Hughes HE, Temple IK, Faravelli
F, Waggoner D, Turkmen S, Cormier-Daire V, Irrthum A, Rahman N, Childhood Overgrowth Collaboration. 2005. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. Am. J. Hum. Genet. 77: 193–204.
Tatton-Brown K, Rahman N. 2007. Sotos syndrome. Eur. J. Hum. Genet. EJHG 15: 264–271.
Villani A, Greer M-LC, Kalish JM, Nakagawara A, Nathanson KL, Pajtler KW, Pfister SM, Walsh MF,
Wasserman JD, Zelley K, Kratz CP. 2017. Recommendations for Cancer Surveillance in Individuals with RASopathies and Other Rare Genetic Conditions with Increased Cancer Risk. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 23: e83–e90.
Waggoner DJ, Raca G, Welch K, Dempsey M, Anderes E, Ostrovnaya I, Alkhateeb A, Kamimura J, Matsumoto
N, Schaeffer GB, Martin CL, Das S. 2005. NSD1 analysis for Sotos syndrome: insights and perspectives from the clinical laboratory. Genet. Med. Off. J. Am. Coll. Med. Genet. 7: 524–533.
Professionnels de santé en Occitanie - Toute anomalie mise en évidence lors du bilan initial et lors du suivi doit être prise en charge par l’équipe médicale spécialisée.
Centre de Référence des Anomalies du Développement et Syndromes Malformatifs : pour le diagnostic et la coordination des prises en charge à tout âge de la vie. Le centre de référence propose une consultation de diagnostic et de suivi spécialisée
Hôpital Arnaud de Villeneuve - CHRU Montpellier
Département de génétique médicale
34295 Montpellier cedex 5
Cardiopédiatrie : pour le suivi des enfants et des adolescents - centre de compétence
Hôpital Arnaud de Villeneuve - CHRU Montpellier
Département de Pédiatrie (Pôle Enfant)
34295 Montpellier cedex 5
Cardiologie adulte : pour le suivi à l’âge adulte Centre de compétence
Hôpital Arnaud de Villeneuve - CHRU Montpellier
Département de Cardiologie et Maladies Vasculaires
34295 Montpellier cedex 5
Chirurgie orthopédique et plastique infantile : pour les scolioses Centre de compétence
Hôpital Lapeyronie - CHRU Montpellier
Département de chirurgie infantile (Pôle Enfant)
34295 Montpellier cedex 5
Endocrinologie pédiatrique : pour les troubles hormonaux Centre de compétence
Hôpital Arnaud de Villeneuve - CHRU Montpellier
Département de Pédiatrie (Pôle Enfant)
34295 Montpellier cedex 5
Gastrologie pédiatrique : pour les difficultés alimentaires et troubles du transit Centre de compétence
Hôpital Arnaud de Villeneuve - CHRU Montpellier
Département de Pédiatrie (Pôle Enfant)
34295 Montpellier cedex 5
Néphrologie pédiatrique : pour les anomalies rénales Centre de référence
Hôpital Arnaud de Villeneuve - CHRU Montpellier
Département de Pédiatrie (Pôle Enfant)
34295 Montpellier cedex 5
Neuropédiatrie et neurochirurgie pédiatrique : pour le suivi développemental et des complications neurologiques Centre de compétence
Hôpital Gui de Chauliac - CHRU Montpellier
Département de Pédiatrie
34295 Montpellier cedex 5
Ophtalmologie pédiatrique : pour le dépistage et la surveillance des troubles de la vue Centre de référence
Hôpital Gui de Chauliac - CHRU Montpellier
Département d’ophtalmologie
34295 Montpellier cedex 5
ORL pédiatrique : pour le dépistage et la surveillance des troubles de l’audition Centre de référence
Hôpital Gui de Chauliac - CHRU Montpellier
Département ORL et chirurgie cervico-faciale (Pôle Neurosciences Tête et Cou)
34295 Montpellier cedex 5
Psychiatrie de l’enfant et de l’adolescent : en cas de troubles du comportement Centre de compétence - Centre du secteur territorial en première intention
SMPEA Peyre Plantade - CHRU Montpellier
34295 Montpellier cedex 5
Urologie pédiatrique : pour les problèmes des voies génito-urinaires Centre de compétence
Hôpital Lapeyronie - CHRU Montpellier
Département de chirurgie infantile (Pôle Enfant)
34295 Montpellier cedex 5