Guide pratique : Ostéogenèse imparfaite
Rédigé par Valentin Ruault, interne de génétique, Dr Fanny Alkar, Dr Cyril Amouroux, Dr Aurélia Carbasse, Dr Marjolaine Willems responsable Centre de Compétence Maladies Osseuses Consitutionnelles (MOC)
Publié Novembre 2020 - Réf. OMIM et ORPHA : voir paragraphe "aspects génétiques"
Qu'est-ce que l'ostéogenèse imparfaite ?
Plusieurs noms ont été donnés à cette maladie, dont les plus connus sont “maladie des os de verre” ou “maladie de Lobstein”. Cela ne signifie pas que l’ostéogenèse imparfaite n’existait pas avant le XVIIIème siècle : des travaux récents ont permis d’identifier chez une momie égyptienne vieille de 3000 ans des signes d’ostéogenèse imparfaite.
Les différentes formes d’ostéogenèse imparfaite sont habituellement divisées en 4 groupes :
• “
• “
• “
• “Ostéogenèse imparfaite intermédiaire avec sclère normale” ou “Ostéogenèse imparfaite de type 4”
Il est habituel de résumer ainsi la sévérité de la maladie :
• Sévérité modérée du type 1
• Sévérité majeure entraînant un décès avant la naissance ou peu de temps après la naissance dans le type 2
• Sévérité importante et progressive du type 3
• Sévérité variable du type 4
Cette classification est utile pour organiser la prise en charge des patients mais il est parfois difficile de prédire avec précision la sévérité de l’atteinte chez un individu donné.
L’objectif de ce guide n’est pas d’être exhaustif mais d’être le plus compréhensible possible. Aussi, pour plus de clarté, nous ne distinguerons pas ces 4 types d’atteinte. Nous parlerons plus simplement du spectre de la maladie, qui s’étend des formes létales aux formes presque sans symptômes (parfois découvertes chez des parents qui n’ont jamais eu de fractures).
Caractéristiques clinico-biologiques
Dans les formes sévères, mais également dans certaines formes légères, des signes cliniques peuvent être vus par échographie chez le fœtus, pendant la grossesse. Il s’agit le plus souvent de raccourcissement, de déformation à type d’incurvation de certains os, de signes de fractures, d’une déformabilité de la boîte crânienne au passage de la sonde d’échographie qui peuvent concerner tous les os. Dans certains cas, le médecin pourra demander à réaliser un scanner osseux du fœtus, qui permettra de mieux voir les os et leurs déformations et donc de préciser la sévérité.
Durant l’enfance, les signes cliniques les plus habituels sont les fractures : elles sont répétées et surviennent avec des traumatismes modérés, parfois même en l’absence de traumatisme selon le degré de sévérité de l’atteinte osseuse. Ces fractures peuvent survenir à tout âge et sur tous les os. Les douleurs osseuses sont fréquentes, même en l’absence de fracture. Certains types de fractures sont plus évocateurs que d’autres d’ostéogénèse imparfaite (fractures précoces, transverses, bilatérales, touchant la partie médiane de l’os - la diaphyse, fractures des vertèbres appelées tassements). Il est important de rappeler que tous les enfants qui présentent des fractures répétées ne souffrent pas d’ostéogenèse imparfaite. Le pédiatre et le généticien recherchent également des arguments pour évoquer d’autres causes, isolées ou associées, comme une carence vitaminique par exemple.
Autres signes cliniques :
• Sclérotique bleutée chez environ 80% des individus (la sclérotique est le nom médical du “blanc de l’œil” - voir photo - source : Wikipédia)
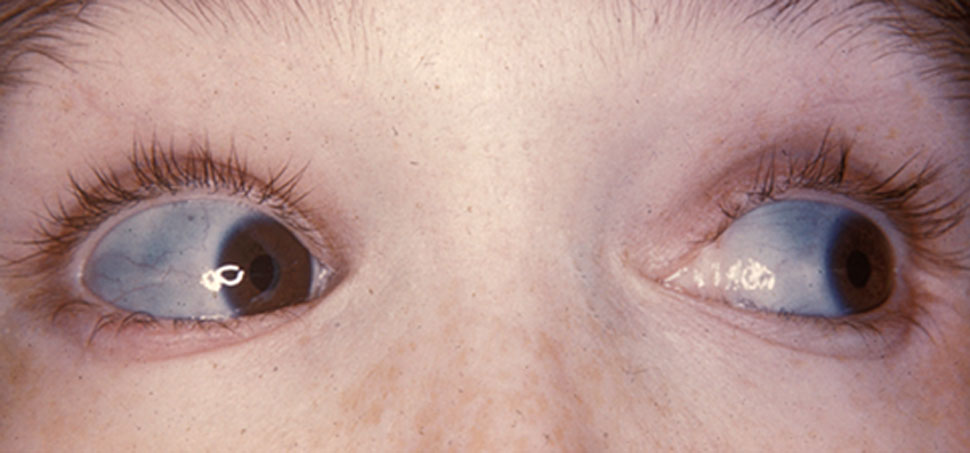

• Hyperlaxité ligamentaire (souplesse excessive des articulations)
• Déformation de certains os (scoliose, pectus carinatum, parfois incurvation des os des jambes)
• Taille inférieure à celle des autres enfants
• Surdité (très rare chez les enfants mais présente à un degré variable chez 40% des adultes)
• Peau légèrement transparente et souple
• Hématomes fréquents, sur tout le corps
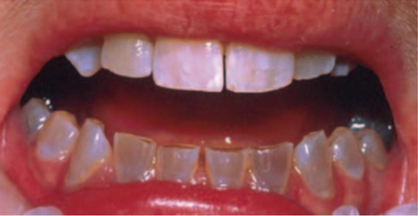
• Dentinogenèse imparfaite : c’est un défaut de formation des dents, qui ne sont pas aussi solides qu’elles devraient l’être, et peuvent être dans certains cas légèrement translucides (voir photo, source : Calvez 2013). On peut avoir une atteinte sévère des dents alors que l’atteinte des os est limitée, ou à l’inverse, ne pas avoir d’atteinte dentaire alors que les os sont plus sévèrement atteints.

À l’âge adulte, les fractures deviennent en général plus rares. En revanche, les problèmes liés à la colonne vertébrale peuvent s'aggraver. Une surdité peut s’installer chez environ 40% des individus après la puberté. Elle est souvent “de transmission” dans un premier temps mais parfois également “de perception” dans un deuxième temps.
L’intelligence des individus atteints d’ostéogenèse imparfaite est normale.
Signes radiologiques
Le médecin peut être amené à prescrire un “bilan radiologique” à un enfant ou un adulte chez qui on suspecte une ostéogenèse imparfaite. Ce bilan comporte des radiographies de plusieurs os, dont au minimum :
• Le crâne, de face et de profil
• La colonne vertébrale, de profil
À ces 3 clichés s’ajoutent le plus souvent des radiographies d’autres os, comme ceux de la jambe, ou du thorax par exemple.
Ces radiographies peuvent montrer :
• Des séquelles de fractures anciennes, parfois passées inaperçues
• Des os plus transparents que la moyenne
• Des os wormiens en excès (ce sont des os surnuméraires au niveau du crâne, qui ne sont ni graves ni douloureux, mais qui peuvent aider au diagnostic)
• Des déformations des os (des os incurvés, ou encore l’extrémité des os - les métaphyses - qui sont élargies, …)
• Rarement chez les enfants mais plus fréquemment chez les adultes : des anomalies des vertèbres (“Codfish” vertebrae, que l’on rencontre également dans d’autres maladies avec ostéoporose).
Le médecin demandera parfois une densitométrie osseuse. C’est un examen qui permet d’évaluer la solidité des os. Il se déroule presque comme un examen de radiologie habituel. Chez certains individus, la densité minérale osseuse sera diminuée. Cet examen n’est pas indispensable au diagnostic mais est utile pour surveiller l’efficacité d’un traitement.
Signes biologiques
Parfois, le médecin prescrira un bilan phosphocalcique. C’est un examen qui nécessite une prise de sang. Ce bilan, qui étudie notamment la vitamine D, le phosphore et le calcium, est classiquement normal dans l’ostéogenèse imparfaite. Il est cependant souvent utile de le vérifier afin d’exclure d’autres diagnostics, qui peuvent ressembler à l’ostéogenèse imparfaite, et de vérifier l’absence de carence en vitamine D, en calcium ou en phosphore qui peut aggraver les symptômes de l’ostéogénèse imparfaite.
Aspects génétiques
L’ostéogenèse imparfaite concerne environ 1 personne sur 10 000.
Les gènes impliqués sont résumés dans le tableau ci-dessous, et chaque situation est ensuite détaillée dans les paragraphes A, B et C.
|
Gène |
Mode de transmission |
N° OMIM |
N° Orphanet |
|
COL1A1 |
Autosomique Dominant |
120150 |
1310 - 314029 - 216796 - 216804 - 2016812 - 216820 - 287 - 230857 - 1899 |
|
COL1A2 |
Autosomique Dominant |
120160 |
314029 - 216796 - 216804 - 216812 - 216820 - 230857 - 1899 - 230851 |
|
IFITM5 |
Autosomique Dominant |
614757 |
216828 |
|
BMP1 |
Autosomique Récessif |
112264 |
314029 - 216812 |
|
CREB3L1 |
Autosomique Récessif |
616215 |
216812 |
|
CRTAP |
Autosomique Récessif |
605497 |
216804 - 216812 - 216820 |
|
FKBP10 |
Autosomique Récessif |
607063 |
216812 - 216820 - 1149 - 2771 |
|
MESD |
Autosomique Récessif |
607783 |
- |
|
P3H1 |
Autosomique Récessif |
610339 |
216804 - 216812 |
|
PPIB |
Autosomique Récessif |
123841 |
216804 - 216812 - 216820 |
|
SERPINF1 |
Autosomique Récessif |
172860 |
216812 - 216820 |
|
SERPINH1 |
Autosomique Récessif |
600943 |
216812 |
|
SP7 |
Autosomique Récessif |
606633 |
216820 |
|
SPARC |
Autosomique Récessif |
182120 |
216820 |
|
TENT5A |
Autosomique Récessif |
611357 |
- |
|
TMEM38B |
Autosomique Récessif |
611236 |
216820 |
|
WNT1 |
Autosomique Récessif |
164820 |
216812 - 216820 |
|
MBTPS2 |
Lié à l'X |
300294 |
659 - 2340 - 85284 - 2273 |
A- Autosomique Dominant
Dans 90% des cas, il s’agit d’une affection autosomique dominante, due à un variant pathogène (ou mutation) du gène COL1A1, COL1A2 ou IFITM5.
Dans le cadre de ces formes dites “autosomiques dominantes”, dans un peu plus de 50% des cas, le variant pathogène (ou mutation) du gène en cause est survenu de novo. Cela signifie que le variant pathogène (ou mutation) provient d’un “accident génétique” qui a eu lieu dans l’ovule ou le spermatozoïde avant la fécondation, donc avant que l’embryon ne commence à se former.
Dans ce cas, le risque pour les parents d’avoir un 2ème enfant atteint est faible. Il persiste néanmoins un léger surrisque par rapport à la population générale, estimé autour de 5%, car il est possible qu’un des parents soit porteur d’une mosaïque germinale. Cela signifie que plusieurs spermatozoïdes ou plusieurs ovules sont porteurs du variant pathogène (ou mutation). Alors, le hasard peut faire que ce soit à nouveau un de ces spermatozoïdes ou ovules porteurs du variant pathogène (ou mutation) qui soit à l’origine d’une nouvelle grossesse.
Dans un peu moins de 50% des cas, le variant pathogène (ou mutation) du gène en cause a été hérité d’un des deux parents. Cela est naturellement plus fréquent dans les formes légères. Pour les parents, cela implique un risque d’avoir un 2ème enfant atteint de 50% (voir schéma ci-dessous). Cela peut également être la cause d’une fragilité osseuse connue ou inconnue chez le parent porteur du variant pathogène (ou mutation).
Que le variant pathogène (ou mutation) soit hérité ou apparu de novo, le risque pour l’enfant atteint de transmettre ce variant pathogène (ou mutation) à ses futurs enfants est de 50%.
B- Autosomique récessif
Dans 10% des cas, il s’agit d’une affection autosomique récessive, due à un variant pathogène (ou mutation) d’un de ces gènes : BMP1, CREB3L1, CRTAP, FKBP10, MESD, P3H1, PPIB, SERPINF1, SERPINH1, SP7, SPARC, TENT5A, TMEM38B ou WNT1.
Dans cette situation, pour que l’enfant présente la maladie, il faut que les deux copies d’un même gène portent un variant pathogène (ou mutation). Une des mutations est héritée du père, et l’autre est héritée de la mère. Cela implique un risque pour les parents d’avoir un 2ème enfant atteint de 25% (voir schéma ci-dessous). Les parents, qui sont porteurs d’une mutation sur une seule copie du gène, n’ont pas de symptômes.
L’enfant n’aura un risque de transmettre la maladie à sa descendance que si son ou sa partenaire est également porteur d’un variant pathogène (ou mutation) dans le même gène, ce qui est très rare si le ou la partenaire n’est pas issu de la même famille.
C- Forme liée au chromosome X
Extrêmement rarement, il s’agit d’une affection dont la transmission est liée au chromosome X, due à un variant pathogène (ou mutation) du gène MBTPS2 qui est situé sur le chromosome X. Les femmes ont 2 chromosomes X. Les hommes ont 1 chromosome X et 1 chromosome Y. Seul le chromosome X porte le gène MBTPS2. Dans ce cas, les femmes qui ont 1 copie du gène avec la mutation et 1 copie saine du gène, sans la mutation, ne semblent pas avoir de symptôme. Ce 2ème chromosome X joue le rôle de “roue de secours” qui permet à l’organisme de fonctionner normalement avec uniquement une copie normale du gène. Les hommes, qui ont une copie du gène avec un variant pathogène (ou mutation), sont atteints (voir schéma ci-dessous).
Le chromosome X des garçons est toujours reçu de leur mère, car leur père transmet le chromosome Y.
Le variant pathogène (ou mutation) peut être apparu de novo. Cela signifie que la mutation provient d’un “accident génétique” qui a eu lieu dans l’ovule avant la fécondation, donc avant que l’embryon ne commence à se former. Dans ce cas, le risque pour les parents d’avoir un 2ème enfant atteint est très faible. Il persiste néanmoins un léger surrisque par rapport à la population générale, estimé autour de 5%, car il est possible que la maman soit porteuse dans ses ovules d’une mosaïque germinale. Cela signifie que plusieurs ovules sont porteurs du variant pathogène (ou mutation). Alors, le hasard peut faire que ce soit à nouveau un de ces ovules porteurs de la mutation qui soit à l’origine d’une nouvelle grossesse.
Le variant pathogène (ou mutation) peut également avoir été hérité de la mère. Dans cette situation, la mère n’a pas, ou très peu de symptômes, du fait de son 2ème chromosome X qui porte une copie saine du gène. Dans ce cas, les parents ont une chance sur 4 de donner naissance à une fille non porteuse de la mutation, qui ne présente pas de symptôme et n’aura pas de risque de transmettre la pathologie à sa propre descendance. Ils ont une chance sur 4 de donner naissance à une fille porteuse de la mutation, qui habituellement ne présentera pas de symptôme mais aura un risque de transmission à sa propre descendance. Ils ont une chance sur 4 de donner naissance à un garçon non porteur de la mutation, qui sera indemne de la pathologie et n’aura pas de risque de la transmettre à sa descendance. Ils ont enfin un risque sur 4 de donner naissance à un garçon atteint.
En résumé, pour les parents, le risque d’avoir un 2ème enfant atteint est de 25% : 50% de risque si c’est un garçon, 0% de risque si c’est une fille (voir schéma ci-dessous).
Le risque pour le garçon atteint de transmettre la maladie à ses fils est nul, car il leur transmettra son chromosome Y. Le risque pour le garçon atteint de transmettre la maladie à ses filles est nul, car elles auront un 2ème chromosome X hérité de leur mère qui sera leur “roue de secours”. Cependant, puisque toutes les filles d’un garçon atteint seront porteuses de la mutation, elles auront à leur tour, une fois en âge d’avoir des enfants, toutes un risque de 25% d’avoir un enfant atteint (50% de risque pour un bébé masculin et 0% de risque pour un bébé féminin).
Explication génétique
Nous possédons tous 46 chromosomes. Lors de la conception d’un embryon, et donc d’un enfant, la maman donne un lot de 23 chromosomes, et le papa donne aussi un lot de 23 chromosomes. Parmi ces chromosomes, 22 paires sont identiques chez les femmes et les hommes, numérotées de 1 à 22. On les appelle les autosomes. La dernière paire concerne les chromosomes sexuels. Les femmes ont 2 chromosomes X tandis que les hommes ont 1 chromosome X et 1 chromosome Y.
Pour chaque gène situé sur un autosome (chromosome n°1 à 22), chaque parent porte 2 copies. Il y a donc 4 combinaisons possibles de patrimoine génétique pour l’enfant, schématisées ci-dessous.
Situation AUTOSOMIQUE DOMINANTE
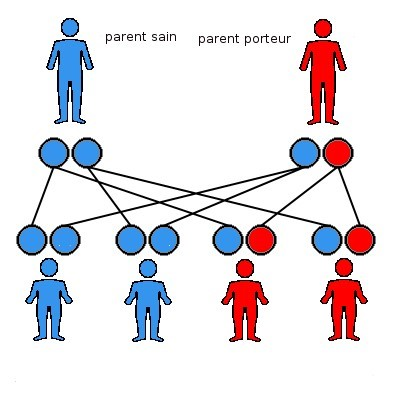

Dans le cadre d’une maladie génétique autosomique dominante, le parent porteur du variant pathogène (ou mutation) a un risque sur deux de transmettre le variant pathogène (schématisée en rouge). L’enfant a donc un risque sur deux d’être porteur du variant pathogène (ou mutation), et de la maladie. Par contre, il n’est pas possible de prédire la sévérité ni le degré d’atteinte de la maladie qui est souvent différente entre deux individus, même porteurs de la même mutation génétique. Une maladie génétique autosomique dominante peut aussi être survenue de novo et donc ne pas avoir été héritée.
Situation AUTOSOMIQUE RÉCESSIVE
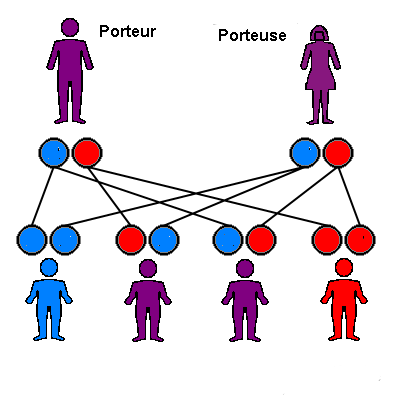
Dans le cadre d’une maladie génétique autosomique récessive, les deux parents sont porteurs d’une mutation (schématisée en rouge). Chacun a un risque sur deux de transmettre la mutation. L’enfant a donc une chance sur quatre de n’être porteur d’aucune mutation (en bleu), a deux risque sur quatre (½) d’être porteur d’une seule mutation (en violet), et un risque sur quatre (25% de risque) d’être porteur des 2 mutations, et donc de présenter la maladie. La sévérité de la maladie est plus souvent comparable entre deux individus porteurs des mêmes mutations génétiques.
Situation LIÉE AU CHROMOSOME X - Si le père est porteur du variant pathogène (ou mutation)
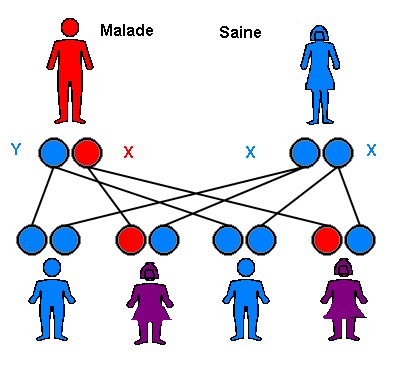
Dans le cadre d’une maladie génétique liée au chromosome X, et si le père est porteur du variant pathogène (ou mutation), alors toutes ses filles seront porteuse d’une copie mutée et d’une copie non mutée. Elles ne seront donc pas atteintes mais “porteuses saines” du variant pathogène (ou mutation). Aucun de ses fils ne sera porteur car il leur aura transmis son chromosome Y. Le risque total d’avoir un enfant atteint est de 0%.
Situation LIÉE AU CHROMOSOME X - Si la mère est porteuse du variant pathogène (ou mutation)
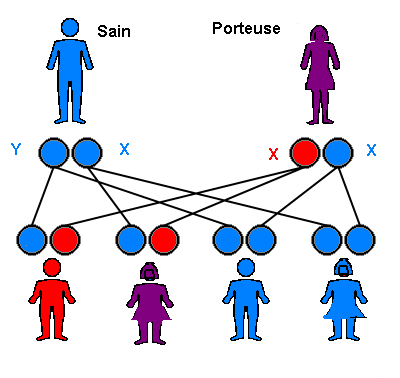
Dans le cadre d’une maladie génétique liée au chromosome X, et si la mère est porteuse du variant pathogène (ou mutation), alors le risque d’avoir un garçon atteint est de 50%, et le risque d’avoir une fille porteuse de la mutation (“porteuse saine”) est de 50%. Le risque global d’avoir un enfant atteint est de 25%. Une maladie génétique liée à l’X peut être survenue de novo et donc ne pas avoir été héritée.
Diagnostic
Lors d’une consultation en génétique, le médecin généticien peut parfois suspecter une ostéogenèse imparfaite. En fonction de l’histoire médicale du patient, de l’histoire médicale de sa famille, de l’examen clinique et de radiologies des os, le diagnostic d’ostéogenèse imparfaite pourra être posé. Il s’agit donc avant tout d’un diagnostic clinique, pour lequel l’analyse génétique n’est ni indispensable, ni systématique.
Dans un second temps, qui n’est pas obligatoire, il peut être proposé d’étudier les gènes dans l’ADN contenu dans les globules blancs du sang ou dans la salive. Il suffira de réaliser une prise de sang ou un frottis jugal, sans nécessité d’être à jeun. Le généticien propose le plus souvent une étude de plusieurs gènes chez le patient (étude d’un panel de gènes, plus rarement étude des séquences codantes de tous les gènes : exome ou étude de tout le génome). S’il s’avère qu’un variant pathogène (ou mutation) dans un des gènes responsables d’ostéogenèse imparfaite est identifié, le diagnostic d’ostéogenèse imparfaite est confirmé et le conseil génétique adapté peut être proposé, en fonction du mode de transmission du gène identifié.
Si un variant pathogène (ou mutation) est mis en évidence chez le patient, le généticien propose habituellement d’effectuer un test génétique chez les parents, afin de savoir si le variant pathogène (ou mutation) est hérité ou de novo (c’est à dire “accidentel”). De plus en plus souvent, il est proposé d’emblée un prélèvement à l’enfant et à ses 2 parents car l’analyse génétique des parents peut aider à l’interprétation de l’analyse chez l’enfant.
L’ostéogenèse imparfaite est une maladie dont la sévérité varie énormément entre les individus. La sévérité peut être si grande que dans certains cas, l'ostéogenèse imparfaite peut entraîner le décès d’un fœtus atteint avant même sa naissance. A l’inverse, certains patients présentant une forme légère n’auront pas de fracture.
Il est fréquent de voir dans une même famille des individus avoir des symptômes de sévérité variable alors qu’ils sont porteurs du même variant pathogène (ou mutation) génétique. Cette variabilité a des limites : un parent atteint de forme légère ne donne habituellement pas naissance à un enfant ayant une forme sévère.
À propos des fractures, il est habituel de dire que :
Dans les formes d’ostéogenèse imparfaite “classiques”, la première fracture survient autour de l’âge d’un an : après que l’enfant a appris à marcher et qu’il commence à tomber. Jusqu’à la puberté, les enfants ont quelques fractures par an, et ce nombre de fracture devient moins important à partir de la puberté. Le nombre de fractures augmente en général à nouveau après 50 ans, surtout chez les femmes à cause de la ménopause. Ces fractures guérissent en général correctement et ne laissent pas de séquelles.
Dans les formes d’ostéogenèse imparfaite “progressivement déformantes”, la première fracture survient plus tôt, en général dès les premières semaines. Les déformations osseuses, si elles touchent les côtes, auront un impact majeur sur le développement des poumons et pourront entraîner des difficultés respiratoires. Les déformations des os des jambes entrainent des difficultés à la marche qui peuvent parfois obliger l’enfant à se servir d’un fauteuil roulant. La taille adulte sera en générale bien inférieure à la taille moyenne des autres adultes.
Conseil génétique
Grossesse :
Pour les femmes atteintes d’ostéogenèse imparfaite de sévérité modérée à sévère, il faudra suivre la grossesse dans une maternité spécialisée, le plus souvent au CHU. En effet, la grossesse chez une femme atteinte d'ostéogenèse imparfaite est plus à risque que les grossesses dans la population générale. Elle peut entraîner notamment une augmentation importante des douleurs, la survenue de fractures. Parfois, la déformation des os au niveau du bassin ne permettra pas l’accouchement par voie naturelle et il faudra alors recourir à une césarienne.
Il est important, dès le début de la grossesse (et idéalement avant le début de la grossesse), de vérifier et d’adapter les apports en calcium, phosphore et en vitamine D, qui sont très importants pour les os. Il suffit le plus souvent d’augmenter la consommation de laitages (plusieurs par jour) et de quelques gouttes de vitamines D chaque jour.
L’allaitement est possible,. Avant la naissance de l’enfant, il faut anticiper l’allaitement avec le médecin gynécologue ou généticien. Il sera alors nécessaire de contrôler l’apport en calcium, phosphore et en vitamine D de la maman.
Il est important de préciser que dans la grande majorité des cas, la grossesse chez une femme atteinte d’ostéogenèse imparfaite se passe bien.
Possibilité de diagnostic prénatal ou de diagnostic pré-implantatoire :
Certains couples à risque d’avoir un enfant atteint d’ostéogenèse imparfaite, souhaitent éviter la transmission de la maladie à leurs futurs enfants. Il existe deux procédures le permettant : le diagnostic prénatal (DPN) ou de diagnostic pré-implantatoire (DPI). Les conditions indispensables à ces deux procédures sont :
1/ de connaître de façon très précise le variant pathogène (ou mutation) génétique en cause de la maladie dans la famille (il faut avoir le résultat du test génétique).
2/ que la demande du couple ait été préalablement discutée et acceptée par un Centre Pluridisciplinaire de Diagnostic PréNatal (CPDPN). En effet, ces deux procédures sont réservées en France aux pathologies jugées comme incurables et d’une particulière gravité. Ce type de procédure n’est donc pas envisagée pour l’ensemble des patients atteints d’ostéogenèse imparfaite, notamment ceux présentant une forme légère.
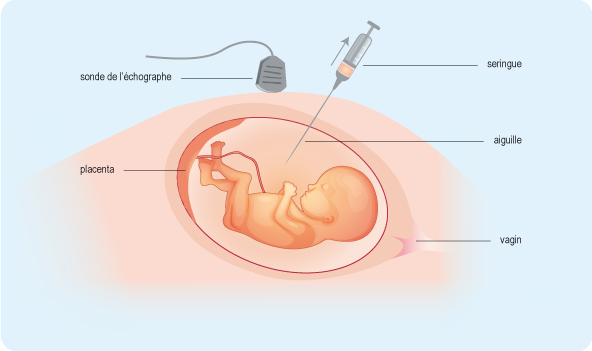
Dans le cadre du diagnostic prénatal, après le début de la grossesse spontanée, le gynécologue va prélever un petit morceau de placenta ou une petite quantité de liquide amniotique à travers le ventre de la maman, en général vers la fin du 3e ou du 4e mois de grossesse (voir schéma). Ce prélèvement pourra être analysé au laboratoire de génétique, habituellement en une semaine, et le couple saura si le fœtus est atteint de la maladie. Le couple pourra alors demander une interruption médicale de grossesse si le fœtus est atteint.
Dans le cadre du diagnostic pré-implantatoire, bien avant de concevoir le bébé, le couple rencontre une équipe de médecins. Les deux parents doivent faire une prise de sang, qui permettra au laboratoire de préparer techniquement la mise au point technique spécifique pour le couple. Le prélèvement d’autres membres de la famille, le plus souvent les futurs grands-parents, sera le plus souvent également nécessaire. La technique consistera à effectuer parallèlement un recueil de spermatozoïdes du père, et, après stimulation hormonale, un recueil d’ovocytes de la mère. Ceci permet de réaliser une fécondation in vitro par injection d’un spermatozoïde dans chaque ovule recueilli (FIV-ICSI). Trois jours plus tard, l’embryon est formé d’environ 8 cellules. On prélève alors une cellule de cet embryon, dont on analyse l’ADN pour y rechercher la mutation familiale. Si l’embryon testé n’est pas porteur de la mutation, on peut le transférer dans l’utérus de la mère. Le fait de recueillir plusieurs ovocytes, et donc de multiplier le nombre d’embryon, augmente les chances d’avoir au moins un embryon qui n’est pas porteur de la mutation. Cette technique permet habituellement d’éviter le recours au diagnostic prénatal mais requiert 18 à 24 mois à partir de la demande du couple pour être mise en œuvre.
Retrouvez plus d’informations sur le diagnostic pré-implantatoire sur ce document : https://www.agence-biomedecine.fr/IMG/pdf/2015_brochure_dpi_vdef.pdf
Prise en charge et traitements
Le premier traitement de l’ostéogenèse imparfaite repose sur la prévention des fractures. Les parents d’un enfant atteint d’ostéogenèse imparfaite doivent apprendre dès l’annonce du diagnostic les mesures de précaution : mobilisation douce et adéquate d’un bébé, habillage avec des vêtements amples, éviter certaines activités à risque (trampoline, sports de contact…). L’activité physique est néanmoins indispensable à la bonne croissance des os et à leur « solidité », des articulations, des muscles. Il faudra donc encourager l’enfant à la pratique d’un sport non traumatique, et à garder par la suite ses bonnes habitudes sportives toute sa vie.
Il est important de prendre en compte toute douleur inhabituelle et de consulter un médecin en cas de doute de fracture. Ces règles de bonne pratique doivent bien sûr se poursuivre tout au long de la vie de la personne atteinte d’ostéogenèse imparfaite et ne pas s'arrêter après l’enfance.
Les apports en calcium, en phosphore, protéines et en vitamine D doivent être adéquats, car ce sont les éléments indispensables à la vie des os, à tous les âges de la vie. On trouve du calcium dans le lait, les yaourts et le fromage ; du phosphore et des protéines dans les laitages, les viandes et poissons. La vitamine D nécessitera toujours un apport supplémentaire à l’alimentation (ampoules de vitamines D à prendre tous les 2 à 3 mois ou quelques gouttes tous les jours).
Les parents doivent disposer à la maison de médicaments anti-douleurs, prêts à être administrés à leur enfant en cas de fracture. Ces médicaments peuvent être du paracétamol, de l'ibuprofène, de la codéine, du tramadol, de la morphine. Un protocole de prise en charge de la douleur en urgence sera proposé par l’équipe du centre de référence ou de compétence maladies osseuses constitutionnelles. En cas de situation d’urgence, il faudra toujours appeler le SAMU en composant le 15 sur son téléphone. Une carte de soins d’urgence, comme celle disponible sur ce site, pourra aider l’équipe médicale à la bonne prise en charge de l’enfant :
https://www.orpha.net/consor4.01/www/cgi-bin/Disease_Emergency.php?lng=FR&stapage=FICHE_URGENCE_O1
La conduite à tenir en cas de fracture doit être apprise aux parents très précocement. Des tutos sont disponibles sur le site OSCAR qui permettent d’apprendre par exemple à immobiliser une fracture avant l’arrivée des secours : https://filiere-oscar.fr/index.php?id=71 ou https://www.youtube.com/channel/UCz1PsLVv7-YJd_HjxNq4xZQ, une vidéo explicative d’une immobilisation « faite maison » d’une fracture.
L’orientation vers un orthopédiste peut être nécessaire selon les déformations osseuses existantes et/ou pour prévenir certaines fractures. La prise en charge orthopédique des fractures diffère de celle des patients ne présentant pas d’ostéogenèse imparfaite. L’immobilisation plâtrée est généralement plus courte. L’enclouage télescopique préventif des fractures est envisagé en fonction du degré de déminéralisation, de la déviation de l’axe des os longs et de la survenue de fractures récurrentes. Les tassements vertébraux et l’apparition d’une scoliose peuvent nécessiter la mise en place d’une rééducation kinésithérapique, d’un corset, ou une intervention chirurgicale pour arthrodèse postérieure dans les cas les plus complexes. Les pieds plats peuvent nécessiter la mise en place d’orthèses plantaires, habituellement après l’âge de 5 ans.
Il pourra être proposé aux parents et à l’enfant, en fonction de son âge, un accompagnement par un psychologue, éventuellement lors de séances “en famille”.
La mise en place d’un traitement par médicaments dans l’ostéogenèse imparfaite peut avoir plusieurs objectifs :
• Réduire le nombre de fractures
• Diminuer les douleurs
• Optimiser la croissance et la taille adulte du patient
• Prendre en charge les comorbidités
Un traitement par bisphosphonates peut être discuté chez les enfants et les adultes atteints d’ostéogenèse imparfaite. Il peut aider à solidifier l’os et donc à réduire le nombre de fractures. Ce traitement ne sera pas indiqué chez tous les enfants : l’équipe médicale décidera, au cas par cas, de l'intérêt ou non de ce traitement.
Le traitement par hormone de croissance n’est habituellement pas indiqué chez les enfants atteints d’ostéogenèse imparfaite. Il peut être utilisé dans certains afin d’améliorer la taille et la solidité osseuse.
La prise en charge de la surdité, si elle est présente, dépendra du type d’atteinte et de sa sévérité. Il pourra s’agir de la mise en place d’un appareillage ou d’une opération chirurgicale.
La prise en charge de la dentinogenèse imparfaite doit se faire en lien avec le centre de référence ou de compétence dédié aux dents. Une hygiène dentaire irréprochable, avec un brossage des dents après chaque repas, est la base de la prise en charge de la dentinogenèse imparfaite.
Fiches de suivi
Scolarisation
La mise en collectivité des enfants atteints d’ostéogenèse imparfaite n’est pas contre-indiquée et ne doit pas être retardée. Elle devra néanmoins être accompagnée avec l’équipe du centre de référence ou de compétences maladies osseuses constitutionnelles afin de limiter au maximum les risques pour l’enfant. Un projet d’accueil individualisé (PAI) pourra être mis en place avec l’équipe médicale et l’équipe scolaire (plus d’informations sur le PAI :
https://www.service-public.fr/particuliers/vosdroits/F21392.
Il faudra également préparer l’accueil à l’école avec l’équipe pédagogique qui mettra en place les adaptations nécessaires en concertation avec le médecin référent (voir http://www.tousalecole.fr/content/ost%C3%A9ogen%C3%A8se-imparfaite-fragilit%C3%A9-osseuse-bep)
Accès aux droits
Des aides financières, telle que l’Allocation pour l’Education des Enfants en situation de Handicap (AEEH), peuvent également être mises en place pour compenser la prise en charge du handicap. L’AEEH est basée sur les constatations des professionnels médicaux et paramédicaux d’une part, et des aidants principaux d’autre part.
Le médecin référent ou le médecin traitant se charge de remplir la demande de prise en charge à 100% au titre d’une Affection Longue Durée (ALD) hors liste. Il remplit également le certificat médical pour le dossier de la Maison Départementale de l’Autonomie (MDA) (anciennement appelée Maison Départementale des Personnes en situation de Handicap, MDPH).
Enfin, le réseau Maladies Rares Méditerranée est également à votre disposition pour vous accompagner dans les différentes démarches sociales : aide à la constitution du dossier pour la Maison Départementale de l’Autonomie (MDA), etc.
Association de malades
Association de l’Ostéogenèse Imparfaite – AOI : https://www.aoi.asso.fr/
Références
Marielle Calvez. La dentinogénèse imparfaite, diagnostic et prise en charge. Autre [q-bio.OT]. 2013. ffdumas-00844555
Lowenstein, Eve J. 2009. “Osteogenesis Imperfecta in a 3,000-Year-Old Mummy.” Child’s Nervous System. https://doi.org/10.1007/s00381-009-0817-7.
Orphanet https://www.orpha.net//consor/cgi-bin/OC_Exp.php?Lng=FR&Expert=666
OSCAR, la filière de santé des maladies rares de l’os, du calcium et du cartilage https://www.filiere-oscar.fr/
PNDS Ostéogenèse imparfaite, HAS, 2016. https://www.has-sante.fr/upload/docs/application/pdf/2016-12/pnds_-_osteogenese_imparfaite.pdf.
Steiner, Robert D., and Donald Basel. 2019. “COL1A1/2 Osteogenesis Imperfecta.” In GeneReviews® [Internet]. University of Washington, Seattle.
Zhytnik, Lidiia, Katre Maasalu, Binh Ho Duy, Andrey Pashenko, Sergey Khmyzov, Ene Reimann, Ele Prans, Sulev Kõks, and Aare Märtson. 2019. “De Novo and Inherited Pathogenic Variants in Collagen-Related Osteogenesis Imperfecta.” Molecular Genetics & Genomic Medicine 7 (3): e559.
Professionnels de santé en Occitanie
Centre de compétence Maladies Osseuses Constitutionnelles : pour le diagnostic et la coordination des prises en charge à tout âge de la vie
Hôpital Arnaud de Villeneuve - CHRU de Montpellier
Département de génétique médicale
34295 Montpellier cedex 5
Centre de Référence des Anomalies du Développement et Syndromes Malformatifs
Hôpital Arnaud de Villeneuve - CHRU de Montpellier
Département de génétique médicale
34295 Montpellier cedex 5
Chirurgie orthopédique et plastique infantile : pour les scolioses, anomalies du palais et les problèmes squelettiques
Hôpital Lapeyronie - CHRU de Montpellier
Département de chirurgie infantile (Pôle Enfant)
34295 Montpellier cedex 5
Endocrinologie pédiatrique : pour les différents traitements médicamenteux et la croissance
Hôpital Arnaud de Villeneuve - CHRU de Montpellier
Département de Pédiatrie (Pôle Enfant)
34295 Montpellier cedex 5
Ophtalmologie pédiatrique : pour le dépistage et la surveillance des troubles de la vue - Centre de référence des affections sensorielles génétiques
Hôpital Gui de Chauliac - CHRU de Montpellier
Département d’ophtalmologie
34295 Montpellier cedex 5
ORL pédiatrique : pour le dépistage et la surveillance des troubles de l’audition - Centre de référence des affections sensorielles génétiques
Hôpital Gui de Chauliac - CHRU de Montpellier
Département ORL et chirurgie cervico-faciale (Pôle Neurosciences Tête et Cou)
34295 Montpellier cedex 5
Psychiatrie de l’enfant et de l’adolescent : en cas de troubles comportementaux - Centre du secteur territorial en première intention
SMPEA Peyre Plantade - CHRU de Montpellier
34295 Montpellier cedex 5
Odontologie pédiatrique : pour la surveillance dentaire
Centre de Soins d'enseignement et de recherche dentaires - CHRU de Montpellier
34295 Montpellier cedex 5