Guide pratique : Syndrome de Rubinstein-Taybi
Qu'est ce que le syndrome de Rubinstein-Taybi ?
Rédigé par : Valentin Ruault et M. le Pr David Geneviève
Le syndrome de Rubinstein-Taybi a été décrit en 1963 et doit son nom aux deux médecins qui ont décrit pour la première fois des patients atteints. Ce syndrome avait été appelé à l’origine le “syndrome des pouces et des gros orteils trop larges”.
Il s’agit d’une anomalie génétique rare caractérisée par un retard de croissance et de développement, ainsi que d’autres signes détaillés plus loin. Elle entraîne la plupart du temps un déficit intellectuel modéré à sévère. Les signes sont variables d’une personne à l’autre.
Les personnes atteintes de Rubinstein Taybi sont souvent décrites comme ayant une personnalité sympathique et joviale.
Origine du syndrome de Rubinstein-Taybi
Le syndrome de Rubinstein-Taybi est une pathologie autosomique dominante. Il est souvent dû à des mutations de novo, c’est à dire accidentelles et non héritées, du gène CREBBP ou du gène EP300. Dans cette situation, le risque pour un couple de parents non malades d’avoir un deuxième enfant atteint du syndrome de Rubinstein-Taybi est légèrement supérieur de celui de la population générale, soit environ 1%. Il existe en effet un risque faible de mosaïque germinale, c’est à dire que l’un des parents puisse être porteur d’une mutation génétique uniquement dans certains de ses ovocytes ou certains spermatozoïdes, et nulle part ailleurs.
En raison de ce risque de mosaïque germinale, un diagnostic prénatal pour une future grossesse peut être réalisé à la demande du couple, si une mutation a été mise en évidence chez le cas-index (le patient atteint). En l’absence de diagnostic prénatal, une étude échographique de la morphologie fœtale par un échographiste référent peut être un élément rassurant pour les parents.
Le risque pour un patient atteint, avec mutation dans l’un de ces deux gènes, d’avoir un enfant avec le syndrome de Rubinstein-Taybi est par contre de 50%.
Explication génétique
Dans ce syndrome, une des copies du gène CREBBP (MIM *600140) est atteinte, et donc peu ou non fonctionnelle. L’autre est normale, mais ne suffit pas à compenser cette perte. On parle alors de pathologie « dominante ». Il est situé sur le chromosome 16. Cette situation représente 40 à 50% des cas.
Il existe un deuxième gène, EP300 (MIM *602700), sur le chromosome 22, qui peut également être responsable du syndrome de Rubinstein-Taybi. Ici encore on parle de pathologie « dominante ». Il n’est impliqué que chez 3 à 8% des patients avec RTS.
Un syndrome proche lui est rattaché : la délétion 16p13.3 (MIM # 610543). En effet, lors de la délétion d’un bout du bras court d’un des deux chromosomes 16, on observe parmi les gènes délétés, CREBBP. Cette situation représente environ 10% des cas.
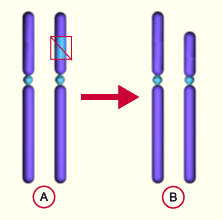

A : Chromosome normal
B : Délétion d’un bout d’un des deux chromosomes
Nous possédons tous 46 chromosomes. Lors de la conception d’un embryon, et donc d’un enfant, la maman donne un lot de 23 chromosomes, et le papa donne aussi un lot de 23 chromosomes. Nous possédons donc un double de chacun de nos chromosomes, et donc un double de chacun de nos gènes. Puisque nous possédons tous deux lots de 23 chromosomes, il y a quatre combinaisons possibles de patrimoine génétique pour l’enfant, schématisées ci-dessous. Chaque rond représente un lot de chromosomes.
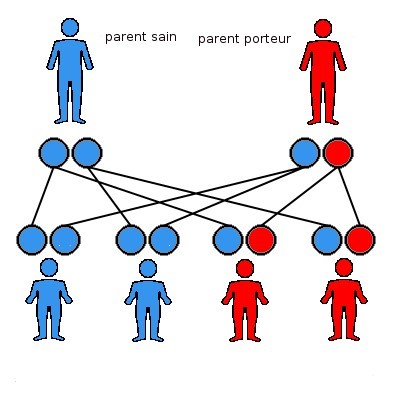

Dans le cadre d’une maladie génétique dominante, le parent porteur de la mutation a un risque sur deux de transmettre la mutation (le lot de chromosomes rouge). L’enfant a donc un risque sur deux d’être porteur de la mutation, et de la maladie. Par contre, il n’est pas possible de prédire la sévérité ni le degré d’atteinte de la maladie qui est souvent différente entre deux personnes ayant la même mutation génétique.
Dans presque tous les cas, les enfants atteints du syndrome de Rubinstein-Taybi ont des parents qui ne sont pas porteurs de la mutation dans le gène CREBBP ou le gène EP300. On dit alors que la mutation est “de novo”. Cela signifie que la mutation est apparue de manière accidentelle au moment de la conception du bébé.
La protéine CREBBP joue un rôle important dans l’acétylation des histones (des protéines qui entourent l’ADN) et donc dans la régulation de l’activité de nos gènes. Elle est active dans tous les tissus de notre corps, et est, entre autre, indispensable à la division cellulaire lors du développement de l’embryon. La protéine EP300 est également un régulateur de la transcription par modulation de la chromatine (donc elle module l’ADN). Ces deux protéines travaillent ensemble de manière combinée en contrôlant l’acétylation des histones et l’expression des gènes au cours du développement.
Le nombre de personnes atteintes par ce syndrome est difficile à estimer, mais il toucherait environ 1 naissance sur 125 000 (référence : Orphanet).
L’addition de ces trois phénomènes (mutation du gène CREBBP, du gène EP300 ou délétion du chromosome 16) n’explique pas 100% des patients. Il reste en effet environ 30% des patients pour lesquels aucune explication génétique n’est trouvée. Le diagnostic est alors exclusivement posé par les symptômes cliniques évocateurs du RTS. Dans ce cas, il n’est pas possible de proposer un diagnostic prénatal ou préimplantatoire pour une future grossesse.
Diagnostic
Lors d’une consultation en génétique, le médecin généticien peut parfois suspecter un syndrome de Rubinstein-Taybi. Il est alors proposé d’étudier les gènes CREBBP et EP300 dans l’ADN contenu dans les globules blancs ou dans la salive. Il suffira de réaliser une prise de sang ou un frottis jugal, sans nécessité d’être à jeun.
Si les symptômes cliniques ne sont pas évocateurs du RTS, le généticien propose une étude de plusieurs gènes chez le patient (un panel de gènes ou un exome). S’il s’avère qu’une mutation dans le gène CREBBP ou le gène EP300 est retrouvée, le diagnostic de RTS est confirmé. On peut également identifier une délétion 16p13.3, grâce à un examen s’appelant ACPA (Analyse Chromosomique par Puces à ADN), qui peut également expliquer les symptômes.
Si une mutation ou une délétion est mise en évidence chez le patient, le généticien propose d’effectuer un test génétique chez les parents, afin de savoir si la mutation ou la délétion est héritée ou de novo (c’est à dire “accidentelle”).
Caractéristiques Cliniques
Les caractéristiques cliniques du syndrome de Rubinstein-Taybi (avec entre parenthèses le pourcentage de patients touchés par le symptôme) sont les suivantes :
- une déficience intellectuelle (définie par un QI inférieur à 70) très fréquente mais de degré variable : le QI moyen se situe autour de 50, mais peut varier entre 24 et 80 ;
- des troubles du comportement (très fréquents) ;
- des troubles de l’attention (très fréquents) ;
- un retard de croissance en taille et en poids avec petit périmètre crânien dès la petite enfance (très fréquent) ;
- des troubles de la vue (80%, détails ci-dessous) ;
- des troubles alimentaires (75%) ;
- des malformations génito-urinaires (52%) ;
- un risque d’obésité débutant avant la puberté chez les garçons, et à l’adolescence chez les filles (40-50%) ;
- des malformations cardiaques (33%) ;
- des crises d’épilepsie (28%) ;
- une surdité modérée (24%) ;
- des troubles du sommeil (moins fréquents) ;
- un risque tumoral plus important que la population générale (mais tout de même rare) ;
- un risque anesthésique légèrement plus important que la population générale.
Il s’y ajoute d’autres symptômes, et quelques particularités morphologiques (voir ci-dessous).
Au niveau des extrémités (très fréquents) :
- des mains et des pieds courts
- des pouces et des gros orteils larges
Manifestations dentaires (très fréquentes) :
- des caries fréquentes
- la présence d’une cuspide surnuméraire des incisives centrales supérieures définitives, sans impact sur le quotidien du patient
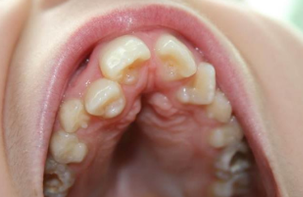

Photo issue du PNDS « Syndrome de Rubinstein-Taybi »
- malocclusion dentaire (fréquence non définie)
Manifestations ophtalmologiques (80%) :
- des conjonctivites (45%)
- un strabisme
- des troubles de la réfraction (myopie, hypermétropie, astigmatisme)
- un ptosis
- un colobome irien et/ou rétinien (rare, c’est une anomalie de développement du cristallin, de l’iris, de la choroïde ou de la rétine ; illustration : https://fr.wikipedia.org/wiki/Colobome#/media/File:Coloboma.jpg)
- une cataracte (rare)
- une hypoplasie du nerf optique (rare)
{kind=link}
Manifestations uro-génitales (52%) :
- une cryptorchidie chez le garçon (des testicules qui ne sont pas complètement descendus dans les bourses, presque 100%)
- hydronéphrose (une dilatation des reins causée par une obstruction des uretères, qui conduisent l’urine des reins à la vessie)
- duplication de l’arbre urinaire
- reflux vésico-urétéral
Manifestations endocrinologiques (fréquence non définie) :
- une petite taille qui apparaît tôt après la naissance malgré des mensurations de naissance dans la moyenne : la taille moyenne des femmes adultes est de 151 cm, et celle des hommes de 163 cm (soit -2 Déviations Standard par rapport à la moyenne pour les hommes comme pour les femmes)
- des hypoglycémies néonatales, voire parfois des hyperglycémies
- un risque d’obésité plus élevé que dans la population générale, débutant dans l’enfance chez les garçons et à l’adolescence chez les filles (40-50%)
- la puberté débute à un âge normal
Manifestations digestives (fréquence non définie) :
- un reflux gastroœsophagien (RGO, fréquent)
- une constipation
Manifestations cardiaques congénitales (33%) :
- communication interventriculaire, communication interauriculaire, persistance du canal artériel, sténose de l’aorte, coarctation de l’aorte, sténose pulmonaire ou bicuspidie aortique
Manifestations de la sphère ORL (fréquence non définie) :
- une surdité de transmission modérée, rarement de perception, favorisée par des otites moyennes aiguës fréquentes (24%)
- un palais creux (rare)
- des apnées du sommeil (rare)
Manifestations neurologiques (fréquence non définie) :
- épilepsie (convulsions pour 28% des patients, et EEG anormal dans 60% des cas) habituellement pharmacosensible et assez facile à traiter.
- agénésie du corps calleux (rare, illustration : https://fr.wikipedia.org/wiki/Corps_calleux#/media/File:Corpus_callosum.gif )
- rétrécissement du cône terminal de la moelle (rare)
{kind=link}
Manifestations squelettiques (rare) :
- une scoliose
- une instabilité fémoro-patellaire (3%)
- une luxation congénitale de hanche (1%)
Manifestations cutanées (fréquence non définie) :
- angiomes capillaires
- hypertrichose
- cicatrices chéloïdes
- pilomatricomes
Il est bien entendu qu’aucun patient ne présente tous ces symptômes, et qu’on ne retrouve chez chacun qu’un nombre variable de ceux-ci.
Focus sur les compétences motrices et cognitives
Les premiers mots apparaissent vers l’âge de 25 mois. Certains enfants restent non verbaux. La marche est généralement acquise vers 30 mois. La propreté est en moyenne acquise lors de la 5e année.
Le QI moyen des patients atteints du syndrome de Rubinstein-Taybi est globalement compris entre 35 et 50, mais peut varier de 24 à 80. Certains patients présentent donc une déficience intellectuelle, définie par un QI inférieur à 70, tandis que d’autres, qui ont un QI supérieur à 70, ont des difficultés d’apprentissage. Le QI moyen se situe autour de 50. Le QI performance est souvent meilleur que le QI verbal, ce qui signifie que les difficultés prédominent sur le langage, alors que la compréhension est en règle générale meilleure.
Les patients atteints du syndrome de Rubinstein-Taybi sont souvent décrits comme ayant une personnalité sympathique et joviale.
Pronostic
La sévérité de la pathologie est variable. Parfois, l’autonomie des patients est possible.
Traitement
Aujourd’hui, il n’existe pas de traitement capable de guérir le syndrome de Rubinstein-Taybi.
On peut en revanche dépister précocement, prévenir ou traiter certains symptômes, par exemple :
- des antiépileptiques, contre les crises d’épilepsie ;
- du méthylphénidate contre les troubles du comportement et de l’attention ;
- de la mélatonine pour favoriser l’endormissement ;
- des hormones de croissance (s’il y a un déficit de ces hormones), pour atteindre une taille adulte plus proche de la moyenne de la population générale ;
- des prothèses auditives, contre la perte d’audition ;
- des lunettes de vue, contre les troubles oculaires ;
- des séances d’orthophonie afin d’améliorer la communication ;
- des anti-acides contre le reflux gastro-oesophagien, voire parfois une opération chirurgicale (fundoplicature de Nissen) ;
- des conseils diététiques pour lutter contre l’obésité.
La chirurgie corrige le plus souvent les malformations. Citons par exemple :
- la chirurgie cardiaque ;
- la cure chirurgicale de cryptorchidie ;
- l’orthodontie en cas de malocclusion dentaire ;
- la chirurgie orthopédique, par exemple en cas de scoliose très sévère, ou de dysplasie des hanches sévère.
ATTENTION : la nature des appareillages auditifs rend souvent leur prise en charge par les CPAM et MDPH difficile. En cas de mauvais remboursement des dépenses vous pouvez demander de l’aide auprès d’une assistante sociale pour remplir votre dossier MDPH. Le Réseau Maladies Rares est à votre disposition pour vous conseiller en cas de difficultés pour les prises en charge et les remboursements des appareillages.
Surveillance
Le dépistage des anomalies suivantes doit être mis en œuvre par des équipes spécialisées.
Toute anomalie mise en évidence lors du bilan initial doit être prise en charge par l’équipe médicale spécialisée. Le centre de référence « anomalies du développement » en charge de la maladie peut vous orienter vers les équipes spécialisées.
La surveillance générale sera programmée par le Généticien puis le Pédiatre ou le Médecin Traitant, avec la famille, en fonction des problèmes médicaux.
Bilan à la découverte du diagnostic
NB : Un Protocole National de Diagnostic et de Soins est disponible pour le Syndrome de Rubinstein-Taybi (https://www.has-sante.fr/portail/upload/docs/application/pdf/2017-09/pnds_syndrome_de_rubinstein-taybi.pdf)
● Examen clinique attentif avec recherche de signes en faveur d’un Reflux Gastro-Œsophagien ou d’une hypertension artérielle, palpation testiculaire, examen des hanches et du rachis
● Consultation en cardiopédiatrie
○ Échocardiographie si conseillée par le cardiopédiatre
○ ECG si conseillé par le cardiopédiatre
● Consultation en neuropédiatrie
○ EEG si conseillé par le neuropédiatre
● Consultation en orthopédie
● Consultation en endocrino-pédiatrie ou endocrinologue adulte
○ Bilan glucidique (glycémie et HbA1c)
● Consultation en ORL
○ Audiogramme
● Consultation en ophtalmologie (lampe à fente + fond d’oeil)
● Consultation chez un chirurgien-dentiste
● Consultation chez un diététicien
● Échographie rénale
● Évaluation de la fonction rénale (ionogramme, urée, créatinine, ionogramme urinaire)
Ne pas oublier :
• 100% ALD hors liste
• dossier MDPH
• orientation CAMSP si nécessaire et si enfant âgé de moins de 6 ans
• PAI et/ou PPS si en âge scolaire
• soutien scolaire si nécessaire
• discuter de l'intérêt d’une AVS
• en fonction de l’âge, discuter l'intérêt d’une notification IME avec l’équipe d’enseignants référents et les parents
Dans l’enfance
● Surveillance clinique :
○ du développement psychomoteur (prévoir un bilan de développement et un bilan psychométrique) et des acquisitions scolaires
○ annuelle de la croissance
○ annuelle de l’apparition d’une HTA par mesure de la tension artérielle
○ d’une puberté précoce (rare)
● Bilan paraclinique :
○ bilan ophtalmologique annuel
○ bilan endocrinien de la croissance si la taille est inférieure à -2 DS
○ si infections ORL à répétition : dosage pondéral des immunoglobulines et NFS
○ bilan auditif annuel
○ bilan dentaire annuel
○ orthodontie si besoin dès les premières dents définitives
○ évaluation diététique des apports caloriques si surpoids
○ évaluation annuelle de la fonction rénale (ionogramme, urée, créatinine, ionogramme urinaire) si atteinte rénale
○ orthophonie si nécessaire
○ psychomotricité si nécessaire
À l’adolescence
● Surveillance clinique :
○ du développement et des acquisitions scolaires
○ annuelle de l’apparition d’une scoliose
○ si scoliose : surveillance semestrielle et consultation en chirurgie orthopédique
○ annuelle de la croissance
○ annuelle de l’apparition d’une HTA par mesure de la tension artérielle
○ consultation en gynécologie pour les femmes après la puberté et discussion de la mise en place d’une contraception
● Bilan Paraclinique :
○ bilan ophtalmologique annuel
○ bilan endocrinien de la croissance si la taille est inférieure à -2 DS
○ bilan auditif annuel
○ bilan dentaire annuel
○ orthodontie si besoin
○ évaluation diététique des apports caloriques dès le début de la puberté
○ évaluation annuelle de la fonction rénale (ionogramme, urée, créatinine, ionogramme urinaire) si atteinte rénale
○ orthophonie si nécessaire
○ kinésithérapie si nécessaire
○ ergothérapie si nécessaire
● Préparer l’orientation et l’aide à l’insertion dans le monde professionnel si cela est possible
● Discuter l’intérêt d’une protection juridique de type curatelle ou tutelle avant la majorité voir fiche mesures de protection
● https://www.service-public.fr/particuliers/vosdroits/N155
À l’âge adulte
● Surveillance clinique :
○ annuelle de l’apparition d’une HTA par mesure de la tension artérielle
○ annuelle du poids et de la taille
○ suivi gynécologique des femmes identique à celui de la population générale
● Bilan Paraclinique :
○ âge osseux si poursuite de la croissance staturale à l’âge adulte
○ bilan ophtalmologique annuel
○ bilan auditif annuel
○ bilan dentaire annuel
○ évaluation diététique des apports caloriques si surpoids
○ évaluation annuelle de la fonction rénale (ionogramme, urée, créatinine, ionogramme urinaire) si atteinte rénale
○ orthophonie si nécessaire
○ kinésithérapie si nécessaire
○ ergothérapie si nécessaire
Fiches de suivi
Références
RTS, MIM # 180849, ORPHANET 783
Association française du syndrome de Rubinstein-Taybi
Lien : http://www.afsrt.com/
Orphanet
Lien : https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=FR&Expert=783
PNDS : Syndrome de Rubinstein-Taybi
Lien : https://www.has-sante.fr/portail/jcms/c_2794382/fr/syndrome-de-rubinstein-taybi
GeneReviews : Rubinstein-Taybi Syndrome
Lien : https://www.ncbi.nlm.nih.gov/books/NBK1526/
Rubinstein JH, Taybi H. Broad thumbs and toes and facial abnormalities : a possible mental retardation syndrome. Am J Dis Child 1963, 105 : 588-608.
Tajir M, Fergelot P, Lancelot G, Elalaoui SC, Arveiler B, Lacombe D, Sefiani A. Germline mosaicism in Rubinstein-Taybi syndrome. Gene. 2013;518:476–8.
Wiley S, Swayne S, Rubinstein JH, Lanphear NE, Stevens CA. Rubinstein-Taybi syndrome medical guidelines. Am J Med Genet A. 2003 Jun 1;119A(2):101-10.
Donatella Milani, Francesca Maria Paola Manzoni, Lidia Pezzani, Paola Ajmone, Cristina Gervasini, Francesca Menni, Susanna Esposito. Rubinstein-Taybi syndrome: clinical features, genetic basis, diagnosis, and management. Ital J Pediatr. 2015; 41: 4.
Beets L, Rodrıguez-Fonseca C, Hennekam RC. 2014. Growth charts for individuals with Rubinstein–Taybi Syndrome. Am J Med Genet Part A 164A:2300–2309
Professionnels de santé en Occitanie
Centre de Référence des Anomalies du Développement et Syndromes Malformatifs : pour le diagnostic et la coordination des prises en charge à tout âge de la vie
Hôpital Arnaud de Villeneuve CHU de Montpellier
Département de génétique médicale
34295 Montpellier cedex 5
Cardiopédiatrie : pour le suivi des cardiopathies de l’enfant et de l’adolescent
Hôpital Arnaud de Villeneuve CHRU de Montpellier
Département de Pédiatrie (Pôle Enfant)
34295 Montpellier cedex 5
Cardiologie adulte : pour le suivi des cardiopathies à l’âge adulte
Hôpital Arnaud de Villeneuve CHU de Montpellier
Département de Cardiologie et Maladies Vasculaires
34295 Montpellier cedex 5
Chirurgie orthopédique et plastique infantile : pour les scolioses, anomalies du palais et les problèmes squelettiques
Hôpital Lapeyronie - CHU de Montpellier
Département de chirurgie infantile (Pôle Enfant)
34295 Montpellier cedex 5
Dermatologie : pour la prise en charge des complications dermatologiques
Hôpital Saint-Eloi – CHU de Montpellier
Pôle Cliniques Médicales - Département de Dermatologie
34295 Montpellier cedex 5
Endocrinologie pédiatrique : pour les troubles de la croissance et de la puberté
Hôpital Arnaud de Villeneuve CHU de Montpellier
Département de Pédiatrie (Pôle Enfant)
34295 Montpellier cedex 5
Gastrologie pédiatrique : pour les difficultés alimentaires et troubles du transit
Hôpital Arnaud de Villeneuve CHU de Montpellier
Département de Pédiatrie (Pôle Enfant)
34295 Montpellier cedex 5
Néphrologie pédiatrique : pour les anomalies rénales
Hôpital Arnaud de Villeneuve - CHU de Montpellier
Département de Pédiatrie (Pôle Enfant)
34295 Montpellier cedex 5
Neuropédiatrie et neurochirurgie pédiatrique : pour le suivi développemental et des complications neurologiques
Hôpital Gui de Chauliac - CHU de Montpellier
Département de Pédiatrie
34295 Montpellier cedex 5
Ophtalmologie pédiatrique : pour le dépistage et la surveillance des troubles de la vue
Hôpital Gui de Chauliac - CHU de Montpellier
Département d’ophtalmologie
34295 Montpellier cedex 5
ORL pédiatrique : pour le dépistage et la surveillance des troubles de l’audition
Hôpital Gui de Chauliac - CHU de Montpellier
Département ORL et chirurgie cervico-faciale (Pôle Neurosciences Tête et Cou)
34295 Montpellier cedex 5
Pédiatrie spécialisée : pour les problèmes endocriniens (taille, puberté)
Hôpital Arnaud de Villeneuve - CHU de Montpellier
Département de Pédiatrie (Pôle Enfant)
34295 Montpellier cedex 5
Psychiatrie de l’enfant et de l’adolescent : en cas de troubles comportementaux
SMPEA Peyre Plantade - CHU de Montpellier
34295 Montpellier cedex 5
Stomatologie pédiatrique : pour la surveillance dentaire
Hôpital Gui de Chauliac - CHU de Montpellier
Département ORL et chirurgie stomatologique (Pôle Neurosciences Tête et Cou)
34295 Montpellier cedex 5
Urologie pédiatrique : pour les problèmes des voies urinaires et cryptorchidies
Hôpital Lapeyronie - CHU de Montpellier
Département de chirurgie infantile (Pôle Enfant)
34295 Montpellier cedex 5